








Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkGE-Portuguese Journal of Gastroenterology
Print version ISSN 2341-4545
GE Port J Gastroenterol vol.26 no.5 Lisboa Oct. 2019
https://doi.org/10.1159/000496094
CLINICAL CASE STUDY
Mauriac Syndrome: A Rare Hepatic Glycogenosis in Poorly Controlled Type 1 Diabetes
Síndrome de Mauriac: Uma glicogenose hepática rara na diabetes tipo 1 mal controlada
Marta Patitaa, Gonçalo Nunesa, António Alves de Matosb, Hélder Coelhoc, Cristina Fonsecaa, Jorge Fonsecaa,b
aHospital Garcia de Orta, Gastroenterology Department, Almada, Portugal; bCiiEM, Centro de Investigação Interdisciplinar Egas Moniz, Monte da Caparica, Portugal; cHospital Garcia de Orta, Pathology Department, Almada, Portugal
* Corresponding author.
ABSTRACT
Background: Hepatic glycogenosis (HG) is a complication of poorly controlled type 1 diabetes mellitus (T1DM), characterized by glycogen accumulation in hepatocytes. Mauriac syndrome (MS) is a glycogenic hepatopathy, initially described in 1930, characterized by growth failure, delayed puberty, cushingoid appearance, hepatomegaly with abnormal liver enzymes, and hypercholesterolemia. HG is a condition with good prognosis and fast resolution after adequate glycemic control (although it has potential for relapse), with no case of evolution to end-stage liver disease described. Case: We describe a 26-year-old female, with T1DM complicated by severe retinopathy. The patient maintained poor glycemic control since childhood, presenting glycated hemoglobin persistently higher than 10% and recurrent episodes of ketoacidosis. In adolescence, she developed hepatomegaly and fluctuating elevation of aminotransferases and triglycerides. A small, nonrepresentative hepatic biopsy suggested macrovacuolar steatosis and mild fibrosis. After 15 years of diabetes, the patient was referred for gastroenterology clinic due to chronic diarrhea and exuberant hepatomegaly. Laboratory showed persistent elevation of aminotransferases and triglycerides. Bilirubin, iron metabolism, and coagulation were normal; viral serologies and autoimmune study were negative. Upper endoscopy, ileocolonoscopy, and enteroscopy presented no lesions. Abdominal magnetic resonance imaging displayed massive hepatomegaly. Liver biopsy was repeated showing marked nuclear glycogenization, mild steatosis, and no fibrosis; electron microscopy revealed very large deposits of glycogen and pleomorphic mitochondria with an unusually dense matrix, described for the first time in this entity. The diagnosis of MS variant and diarrhea due to autonomic neuropathy were assumed. Conclusion: Currently, HG is a well-recognized disease that occurs at any age and can be present without the full spectrum of features initially described for MS. In the era of insulin therapy, this entity represents a rare complication, caused by low therapeutic compliance.
Keywords: Type 1 diabetes mellitus, Hepatic glycogenosis ,Mauriac syndrome, Nonalcoholic fatty liver disease, Liver biopsy
RESUMO
Introdução: A glicogenose hepática (GH) é uma complicação da diabetes mellitus tipo 1 (DM1) mal controlada, caracterizada pela acumulação de glicogénio nos hepatócitos. A síndrome de Mauriac (SM) é uma hepatopatia glicogénica, descrita inicialmente em 1930, caracterizada por atraso de crescimento, puberdade tardia, fácies cushingoide, hepatomegalia, elevação das enzimas hepáticas e hipercolesterolemia. A GH é uma condição com bom prognóstico e com rápida resolução após controlo adequado da glicémia (apesar de ter potencial para recidiva), não havendo descrito nenhum caso de evolução para end-stage liver disease. Caso: Descrevemos o caso de uma mulher de 26 anos, com DM1 complicada por retinopatia grave. A doente manteve inadequado controlo glicémico desde a infância, apresentando hemoglobina glicada persistentemente superior a 10% e episódios recorrentes de cetoacidose. Na adolescência, desenvolveu hepatomegalia, elevação flutuante das aminotransferases e triglicéridos. A biópsia hepática sugeriu esteatose macrovacuolar e fibrose ligeira. Após 15 anos de evolução da diabetes, a doente foi encaminhada para consulta de gastrenterologia por diarreia crónica e hepatomegalia exuberante. Laboratorialmente verificou-se elevação persistente das aminotransferases e dos triglicéridos. A bilirrubina, o metabolismo do ferro e a coagulação eram normais; as serologias virais e o estudo auto-imune foram negativos. A endoscopia digestiva alta, ileocolonoscopia e enteroscopia não apresentavam alterações. A ressonância magnética abdominal mostrou hepatomegalia maciça. A biópsia hepática foi repetida, mostrando glicogenização nuclear acentuada, esteatose leve e ausência de fibrose; a microscopia eletrônica revelou depósitos volumosos de glicogênio e mitocôndrias pleomórficas com uma matriz extraordinariamente densa, descrita pela primeira vez nesta entidade. Foi assumido o diagnóstico de glicogenose hepática no contexto de SM e diarreia devido a neuropatia autonómica. Conclusão: Atualmente, a GH é uma entidade bem reconhecida que pode ocorrer em qualquer idade e pode estar presente sem o espectro completo das características descritas inicialmente para a SM. Na era da insulinoterapia, esta patologia representa uma complicação rara, causada pela baixa adesão à terapêutica.
Palavras-Chave: Diabetes mellitus tipo 1, Glicogenose hepática, Síndrome de Mauriac, Fígado gordo não-alcoólico, Biópsia hepática
Introduction
Hepatic glycogenosis (HG) is a complication of poorly controlled type 1 diabetes mellitus (T1DM), characterized by glycogen accumulation in hepatocytes [1, 2]. HG is one of the components of Mauriac syndrome (MS), initially described in 1930, characterized by growth failure, delayed puberty, cushingoid appearance, hepatomegaly with abnormal liver enzymes, and hypercholesterolemia [2, 3].
The diagnosis of HG includes the exclusion of other causes of liver injury, namely infectious, metabolic, obstructive, or autoimmune diseases [1]. The authors describe a particular case of MS of long evolution, with a repeated histological evaluation separated by 10 years proving no fibrosis worsening.
Case Report
We report the case of a 26-year-old woman, diagnosed with T1DM at 11 years of age, with poor glycemic control since the diagnosis, due to poor therapeutic compliance. During the adolescence, she presented late menarche, hypothyroidism, hepatomegaly, and long-standing dyslipidemia. A severe diabetic retinopathy developed as complication of the T1DM. There were multiple hospital records of emergency admissions due to diabetic ketoacidosis and recurrent episodes of hypoglycemia. Glycated hemoglobin was persistently higher than 10% and hepatomegaly developed, with hypertriglyceridemia (511–861 mg/dL), hypercholesterolemia, and fluctuating elevation of aminotransferases persisting for years. Recorded aminotransferases presented with values as high as aspartate aminotransferase (AST) 1,687 IU/L and alanine aminotransferase (ALT) 1,375 IU/L. When she was 16 years old, a nonrepresentative liver biopsy (only three portal spaces) was obtained and revealed macrovacuolar steatosis, focal nuclear glycogenization, and mild fibrosis. The diagnosis of nonalcoholic steatohepatitis was considered.
At 26 years of age, the patient was referred to the gastroenterology/hepatology outpatient clinic, due to 1 year of fluctuating watery diarrhea and persistent increased liver tests. She was under therapy with levothyroxine 100 mg/day, long-acting insulin at bedtime, and fast-acting insulin at meal times. She denied any use of alcohol, tobacco, or illicit drugs. The clinical examination showed a malnourished patient with a body mass index of 17.8, a cushingoid face, and a major hepatomegaly up to 10 cm below the right costal border.
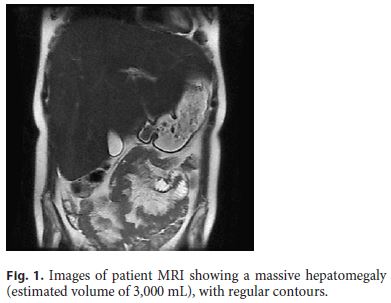
The laboratory data included high glycated hemoglobin (maximum of 14.8%), hypertriglyceridemia (1,066 mg/dL), hypercholesterolemia (224 mg/dL), and hypercortisolemia (25.5 μg/dL). Aminotransferases were high (AST 173 IU/L, ALT 235 IU/L), but gamma-glutamyl transpeptidase and alkaline phosphatase were less than twice the upper normal limit. Bilirubin, iron metabolism, coagulation, and thyroid tests were normal. Viral serology, antibodies for autoimmune liver disorders, and anti-transglutaminase antibody were negative. The genetic study for glycogen storage disease type 1a was also confirmed to be negative. Laboratory evaluation of the diarrhea included the microbiological study of feces, which was negative, as well as the study of serotonin, chromogranin A, gastrin, vasoactive intestinal peptide, glucagon, and urinary 5-hydroxyindoleacetic acid. Ultrasonography confirmed the major hepatomegaly, and CT scan enterography showed no morphological changes in the small bowel. Abdominal magnetic resonance imaging showed massive hepatomegaly (estimated liver volume: 3,000 mL) and a nodular lesion suggestive of focal nodular hyperplasia (Fig. 1). Upper GI endoscopy and ileocolonoscopy were normal, as well as the histology of biopsies taken from the duodenum, colon, and ileum. The capsule enteroscopy presented no mucosal alterations, but the transit in the small intestine was only 2 h.
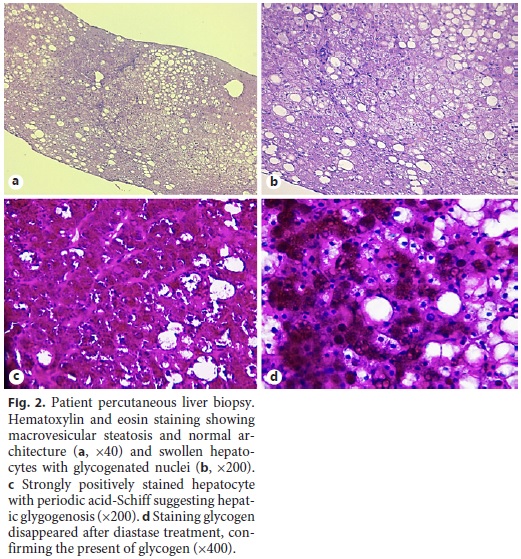
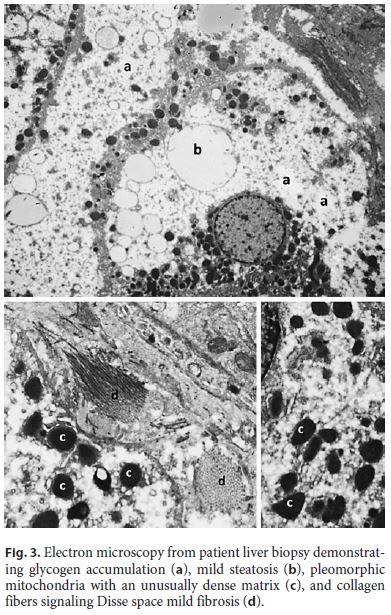
Ten years after the first liver biopsy, a new one was performed, in order to evaluate liver parenchyma and the evolution over this period of time. Optical microscopy revealed intact liver architecture, marked glycogen accumulation and nuclear glycogenization, mild macrovacuolar steatosis, and just a minimal reinforcement of the reticulin network (Fig. 2). In addition, hepatic tissue was also evaluated by transmission electron microscopy, which revealed very large deposits of glycogen and pleomorphic mitochondria with an unusually dense matrix (Fig. 3).
At this point, we established the diagnosis of diabetic glycogenosis in the clinical context of MS and the diagnosis of chronic diarrhea as a consequence of diabetic neuropathy. The importance of improving compliance with glycemic control and adequate diet was reinforced. Therapy with octreotide and loperamide was established. In the reassessment at 3, 6, and 12 months, the patient had less frequent diarrhea and showed a modest improvement in glycemic control, with decrease but incomplete normalization of aminotransferases (2–2.5 times the upper limit of normal values).
Discussion
MS is considered a rare entity in the era of intensive insulin therapy currently prescribed to T1DM patients. However, it still exists and is probably underdiagnosed because of the difficulty in differential diagnosis with nonalcoholic fatty liver disease (NAFLD) [1]. In fact, current evidence shows that NAFLD is rare in T1DM, with a prevalence of less than 10% (lower than in the general population), unlike type 2 diabetes mellitus, in which NAFLD has a prevalence of approximately 70% [4].
Wide fluctuations in plasma glucose levels, with periods of hyperglycemia and hyperinsulinism, appear to be essential to the pathophysiology of HG [2, 5]. High glucose levels promote the flow of glucose into hepatocytes. Then, hyperinsulinemia stimulates glycogen synthase, leading to the conversion of glucose-6-phosphate to glycogen [1, 2]. Furthermore, there is also an increase in serum cortisol levels, as a counter-regulating hormone reactive to hypoglycemia, frequently observed in patients with poorly controlled T1DM [5, 6]. In this patient, poor glycemic control was associated with daily episodes of hypoglycemia, justifying the cushingoid appearance and delayed puberty.
It should also be noted that HG can appear at any age [7]. Initially described in children, there are currently multiple cases described in adults, although not always with the extrahepatic features of MS being present [5, 7, 8]. The patient did not present growth impairment, possibly due to the onset of the T1DM at the age of 11 years and not in very early childhood. In addition to persistently elevated glycated hemoglobin, the presence of microvascular complications translates to prolonged poor control of diabetes, showing that HG may arise in long-term poorly controlled diabetes mellitus and not just in the context of acute ketoacidosis.
Although noninvasive methods are being explored for the diagnosis of HG, such as computed tomography and dual-echo magnetic resonance imaging, liver biopsy is still mandatory for differential diagnosis, namely with NAFLD [1, 9]. The distinction between these two entities is clinically very relevant. In NAFLD, progressive fibrosis and cirrhosis can be expected in a significant number of patients, with a high risk of developing hepatocarcinoma. Conversely, HG presents a favorable evolution, with no evidence of significant fibrosis [1, 10, 11]. The present case demonstrates the good prognosis of HG – two liver biopsies were performed with a 10-year interval, with no evidence of fibrosis worsening, although the aminotransferases have remained floating high during all this period. The authors believe that features of HG were probably present at the age of the first biopsy, and only the scarcity of the biopsy material (three portal spaces) and the rarity of the MS prevented an earlier diagnosis. It should be noted that the diagnosis of HG can be inferred by a therapeutic test of at least 4 weeks, with good glycemic control, allowing to avoid an invasive examination such as liver biopsy. However, if there is doubt in the diagnosis or if adequate glycemic control is not achieved as in the case presented, hepatic biopsy may be considered [11].
In the evaluation of the hepatic parenchyma of patients with glycogenic hepatopathy by transmission electron microscopy, the presence of megamitochondria was previously described, and this feature may be related to the severity of the liver disease [12]. Although no megamitochondria were present in our case, in addition to the large deposits of glycogen, we identified the presence of pleomorphic mitochondria, with an unusually dense matrix, reported for the first time in the MS. These findings reinforce the importance of ultrastructural evaluation, since poor glycemic control and HG may lead to changes in functional level and microanatomy in hepatocytes, as previously described by other authors, and the prognostic importance of these features is not yet fully known. Control of glycemic and insulin doses administered is the basis for the treatment of HG, making possible the complete reversal of hepatomegaly and normalization of transaminases within 2–14 weeks [2].
In conclusion, although it is a rare entity in developed countries, HG should always be considered in a patient with T1DM, poor metabolic control, and hepatomegaly associated with transaminase abnormalities. In the future, the development of noninvasive imaging methods may be useful in the differential diagnosis with NAFLD, but nowadays, hepatic biopsy is still required.
References
1 Giordano S, Martocchia A, Toussan L, Stefanelli M, Pastore F, Devito A, et al. Diagnosis of hepatic glycogenosis in poorly controlled type 1 diabetes mellitus. World J Diabetes. 2014 Dec;5(6):882–8. [ Links ]
2 Julián MT, Alonso N, Ojanguren I, Pizarro E, Ballestar E, Puig-Domingo M. Hepatic glycogenosis: an underdiagnosed complication of diabetes mellitus? World J Diabetes. 2015 Mar;6(2):321–5. [ Links ]
3 Mauriac P. Grosventre, hepatomegalie, troble de la croissance chez les enfants diabetiques: traits depuis plusieursannesparlinsuline. Gaz Hebl Sci Med Bordeaux. 1930;26:402–10. [ Links ]
4 Cusi K, Sanyal AJ, Zhang S, Hartman ML, Bue-Valleskey JM, Hoogwerf BJ, et al. Nonalcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes Obes Metab. 2017 Nov;19(11):1630–4. [ Links ]
5 Hernández-Quiles C, Fernández-Ojeda MR, Solanilla Rodríguez R, Escudero Severin C. [Mauriac syndrome: a liver disease that differs from steatosis of diabetes]. Rev Clin Esp (Barc). 2013 Apr;213(3):169–70. [ Links ]
6 Flotats Bastardas M, Miserachs Barba M, Ricart Cumeras A, Clemente León M, Gussinyer Canadell M, Yeste Fernández D, et al. Hepatomegalia por depósito de glucógeno hepático y diabetes mellitus tipo 1. An Pediatr (Barc). 2007 Aug;67(2):157–60. [ Links ]
7 Parmar N, Atiq M, Austin L, Miller RA, Smyrk T, Ahmed K. Glycogenic Hepatopathy: Thinking Outside the Box. Case Rep Gastroenterol. 2015 Jul;9(2):221–6. [ Links ]
8 Brouwers MC, Ham JC, Wisse E, Misra S, Landewe S, Rosenthal M, et al. Elevated lactate levels in patients with poorly regulated type 1 diabetes and glycogenic hepatopathy: a new feature of Mauriac syndrome. Diabetes Care. 2015 Feb;38(2):e11–2. [ Links ]
9 Sherigar JM, Castro J, Yin YM, Guss D, Mohanty SR. Glycogenic hepatopathy: A narrative review. World J Hepatol. 2018 Feb;10(2):172–85. [ Links ]
10 Abaci A, Bekem O, Unuvar T, Ozer E, Bober E, Arslan N, et al. Hepatic glycogenosis: a rare cause of hepatomegaly in Type 1 diabetes mellitus. J Diabetes Complications. 2008 SepOct;22(5):325–8. [ Links ]
11 Khoury J, Zohar Y, Shehadeh N, Saadi T. Glycogenic hepatopathy. Hepatobiliary Pancreat Dis Int. 2018 Apr;17(2):113–8. [ Links ]
12 Fitzpatrick E, Cotoi C, Quaglia A, Sakellariou S, Ford-Adams ME, Hadzic N. Hepatopathy of Mauriac syndrome: a retrospective review from a tertiary liver centre. Arch Dis Child. 2014 Apr;99(4):354–7. [ Links ]
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to declare.
* Corresponding author.
Marta Patita, MD
Hospital Garcia de Orta, Gastroenterology Department
Av. Torrado da Silva
PT–2805-267 Almada (Portugal)
E-Mail martapatita21@gmail.com
Received: November 20, 2018; Accepted after revision: December 7, 2018
Author Contributions
Drafting of the article: Marta Patita, Gonçalo Nunes. Acquiring of pathology images: Hélder Ceolho, Antóno Alves Matos. Critical revision of the article: António Alves Matos, Hélder Coelho, Cristina Fonseca, Jorge Fonseca. Final approval of the version to be published: Marta Patita, Gonçalo Nunes, António Alves Matos, Hélder Coelho, Cristina Fonseca, Jorge Fonseca.