










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGE-Portuguese Journal of Gastroenterology
versão impressa ISSN 2341-4545
GE Port J Gastroenterol vol.26 no.5 Lisboa out. 2019
https://doi.org/10.1159/000495767
CLINICAL CASE STUDY
Fatty Liver and Autoimmune Hepatitis: Two Forms of Liver Involvement in Lipodystrophies
Fígado gordo e hepatite autoimune: duas formas de atingimento hepático nas lipodistrofias
Andreia Ribeiroa. José Ricardo Brandãob. Esmeralda Cletoc. Manuela Santosd. Teresa Borgese, Ermelinda Santos Silvaa,f
aGastroenterology Unit, Pediatrics Division, Child and Adolescent Department, Centro Materno Infantil do Norte, Centro Hospitalar Universitário do Porto, Porto, Portugal; bAnatomical Pathology Department, Hospital de Santo António, Centro Hospitalar Universitário do Porto, Porto, Portugal; cHematology Unit, Pediatrics Division, Child and Adolescent Department, Centro Materno Infantil do Norte, Centro Hospitalar Universitário do Porto, Porto, Portugal; dNeurology Unit, Pediatrics Division, Child and Adolescent Department, Centro Materno Infantil do Norte, Centro Hospitalar Universitário do Porto, Porto, Portugal; eEndocrinology Unit, Pediatrics Division, Child and Adolescent Department, Centro Materno Infantil do Norte, Centro Hospitalar Universitário do Porto, Porto, Portugal; fInstituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, Porto, Portugal
* Corresponding author.
ABSTRACT
Introduction: Lipodystrophies are a heterogeneous group of rare diseases (genetic or acquired) characterized by a partial or generalized deficit of adipose tissue, resulting in less energy storage capacity. They are associated with severe endocrine-metabolic complications with significant morbidity and mortality. In the pathogenesis of the acquired forms, immunological disorders may be involved. Case 1: A 13-year-old female was diagnosed with acquired generalized lipodystrophy and observed for suspicion of portal hypertension. She presented with generalized absence of adipose tissue, cervical and axillary acanthosis nigricans, and massive hepatosplenomegaly. Laboratory tests revealed AST 116 IU/L, ALT 238 IU/L, GGT 114 IU/L, HOMA-IR 28.2, triglycerides 491 mg/L, and leptin < 0.05 ng/mL. Upper gastrointestinal endoscopy saw no signs of portal hypertension. Hepatic histology showed macrovesicular fatty infiltration (60% of hepatocytes) and advanced fibrosis/cirrhosis. Her clinical conditionworsened progressively to diabetes requiring treatment with subcutaneous insulin and hepatopulmonary syndrome. Case 2: A 15-year-old female, diagnosed with acquired partial lipodystrophy, Parkinson syndrome, autoimmune thyroiditis, and autoimmune thrombocytopenia was observed for hypertransaminasemia since the age of 8 years. She had absence of subcutaneous adipose tissue in the upper and lower limbs and ataxia. Laboratory tests showed AST 461 IU/L, ALT 921 IU/L, GGT 145 IU/L, HOMA-IR 32.6, triglycerides 298 mg/dL, normal leptin levels, platelets 84,000/μL, IgG 1,894 mg/dL, positive anti-LKM and anti-LC-1. Hepatic histology was suggestive of autoimmune hepatitis, without steatosis. She progressed favorably under metformin and immunosuppressive treatment. Conclusion: Early recognition and adequate characterization of liver disease in lipodystrophies is essential for a correct treatment approach. In acquired generalized lipodystrophy, the severe endocrine-metabolic disorder, which leads to steatohepatitis with cirrhotic progression, may benefit from recombinant leptin treatment.
Keywords: Lipodystrophy, Acquired generalized lipodystrophy, Acquired partial lipodystrophy, Fatty liver, Autoimmune hepatitis
RESUMO
Introdução: As lipodistrofias são um grupo heterogéneo de doenças raras (formas genéticas e adquiridas) caracterizadas por défice parcial ou generalizado de tecido adiposo, resultando em menor capacidade de armazenamento energético. Estão associadas a complicações endócrino-metabólicas graves com morbilidade e mortalidade significativas. Na patogénese das formas adquiridas poderão estar envolvidos distúrbios imunológicos. Caso 1: Adolescente de 13 anos, sexo feminino, com diagnóstico de lipodistrofia generalizada adquirida, observada por suspeita de hipertensão portal. Apresentava ausência generalizada de tecido adiposo, acantose nigricans cervical e axilar, e hepatoesplenomegalia volumosa. Do estudo destacavam-se: AST 116 UI/L, ALT 238 UI/L, GGT 114 UI/L, HOMA-IR 28.2, triglicerídeos 491 mg/L e leptina < 0.05 ng/mL. A endoscopia digestiva alta não evidenciou sinais de hipertensão portal. Histologia hepática com esteatose macrovesicular (60% dos hepatócitos) e fibrose avançada/cirrose. A sua condição clínica evoluiu progressivamente para diabetes com necessidade de tratamento com insulina subcutânea e síndrome hepatopulmonar. Caso 2: Adolescente de 15 anos, sexo feminino, com diagnóstico de lipodistrofia parcial adquirida, síndrome parkinsónico, tiroidite autoimune, e trombocitopenia autoimune, observada por elevação das transaminases desde os 8 anos. Apresentava ausência de tecido adiposo subcutâneo nos membros superiores e inferiores, ataxia e tremor das mãos, sem sinais de doença hepática. Do estudo destacavam-se: AST 461 UI/L, ALT 921 UI/L, GGT 145 UI/L, HOMA-IR 32,6, triglicerídeos 298 mg/dL, leptina normal, plaquetas 84,000/μL, IgG 1,894 mg/dL, anticorpos anti-LKM e anti-LC1 positivos. Histologia hepática sugestiva de hepatite autoimune, sem esteatose. A doente evoluiu favoravelmente com metformina e tratamento imunossupressor. Discussão: O reconhecimento precoce e a caracterização adequada da doença hepática nas lipodistrofias são fundamentais para uma correta abordagem terapêutica. Na lipodistrofia generalizada adquirida, o distúrbio endócrino-metabólico grave responsável por esteatohepatite com evolução cirrogénea poderá beneficiar do tratamento inovador com leptina recombinante.
Palavras-Chave: Lipodistrofia, Lipodistrofia generalizada adquirida, Lipodistrofia parcial adquirida, Fígado gordo, Hepatite autoimune
Introduction
Lipodystrophies are a heterogeneous group of rare diseases characterized by partial or complete absence of adipose tissue resulting in the reduction of energy storage capacity with no nutritional deprivation or catabolic state [1]. According to its etiology (genetic or acquired) and the distribution of lost adipose tissue (partial or generalized), they can be categorized into four main categories: congenital generalized lipodystrophy, acquired generalized lipodystrophy (AGL), familial partial lipodystrophy, and acquired partial lipodystrophy (APL) [1, 2].
Lipodystrophies are considered ultra-rare diseases with an estimated prevalence of 1.3–4.7 cases/million, which explains the low awareness of the majority of clinicians for their diagnosis and management [3, 4]. These diseases have a significant impact on the patients life due to endocrine-metabolic morbidity and a psychological impact on body image [1, 5].
Their main features are deficiency in/resistance to leptin because of adipose tissue loss, leading to severe complications such as immunological impairment, hormone dysfunction, and glucose and fat metabolism impairment with ectopic fat accumulation, leading to organ disfunction (mainly liver, kidney, and heart) and premature deaths [6].
The type of liver disease in lipodystrophies is related to their pathophysiology or possible underlying pathogenesis. The authors present two patients with lipodystrophy illustrating both situations.
Case Report
Case 1
A 12-year-old girl was first seen at the Pediatric Hepatology outpatient clinic for voluminous hepatosplenomegaly and suspicion of portal hypertension. Her family history was unremarkable.
The girl had been diagnosed with generalized lipodystrophy at the age of 4 (when changes in body composition were noted) and, at the age of 8 years, she developed physical signs of insulin resistance (acanthosis nigricans). By the age of 10, echocardiography assessment revealed a concentric left ventricular hypertrophy, recommending avoidance of sports. Laboratory tests showed neutropenia and iron deficiency anemia that resolved with oral iron treatment. Despite extensive workup, neutropenia of unknown etiology persisted. At the age of 11, she had hepatosplenomegaly and the possibility of portal hypertension was considered.
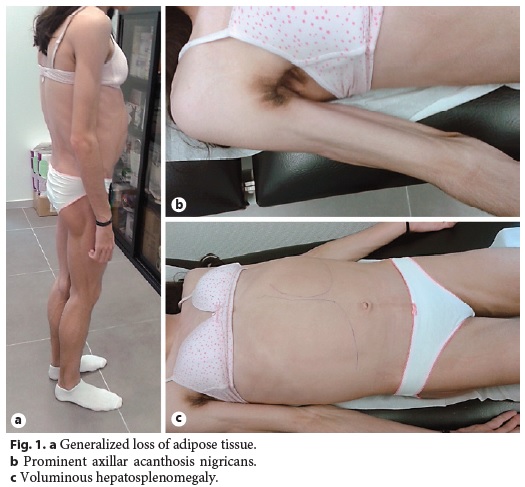
Physical examination revealed generalized loss of adipose tissue with hand and foot involvement (Fig. 1a), cervical, axillary and inguinal acanthosis nigricans (Fig. 1b), and massive hepatomegaly and splenomegaly (Fig. 1c).
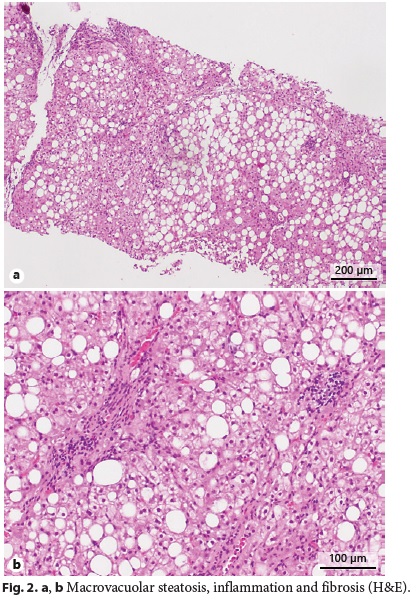
Abdominal ultrasound with Doppler showed an enlarged liver (240 mm) being diffusely hyperechogenic and homogeneous, increased flow velocity of the portal vein (33.1 cm/s), elevation of the hepatic artery Doppler resistance index (0.75), decreased respiratory variation in supra-hepatic veins, and an enlarged spleen (160 mm) with a homogeneous echo pattern. Upper gastrointestinal endoscopy was normal. Liver histology revealed architectural distortion, portal fibrosis with septa, bridges and parenchymal nodularity, macrovesicular hepatocellular steatosis (60% of hepatocytes), ballooning degeneration of hepatocytes, and inflammatory infiltrate composed of lymphocytes (Fig. 2a, b). These findings were consistent with nonalcoholic steatohepatitis diagnosis with advanced fibrosis and progression to cirrhosis.
Laboratory tests revealed an undetectable serum leptin level (< 0.05 ng/mL) with a normal immunological study. Molecular study for genetic lipodystrophies (AGPAT2, BSCL2, PPARG and LMNA genes) was negative and she was classified as AGL.
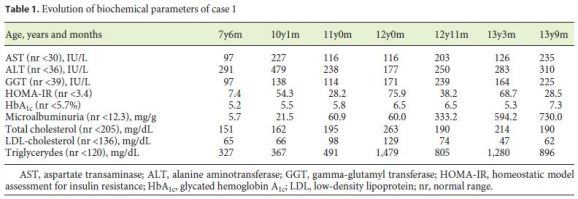
Regarding the evolution of biochemical parameters (Table 1), by 7 years of age, the patient already evidenced raised transaminases and gamma-glutamyl transferase (GGT), increased HOMAIR, and hypertriglyceridemia. At 10 years of age, she started treatment with metformin (500 mg b.i.d.) and enalapril (10 mg/day) because of arising microalbuminuria. There was a transient improvement of her metabolic syndrome, which, however, worsened by the age of 12. Metformin treatment was optimized (1 g b.i.d.) and losartan (50 mg/day) was added due to microalbuminuria exacerbation. Ursodeoxycholic acid (500 mg b.i.d.) and gemfibrozil (600 mg/day) were added, with no significant improvement. Recently, she has been treated with subcutaneous insulin.
Currently, at the age of 13, clinical and biochemical deterioration is evident, with severe hypertransaminasemia, insulin resistance, hypertriglyceridemia, and microalbuminuria. We applied her to the program of early access to treatment with recombinant human leptin, which has recently been authorized. In the last assessment, she had a transcutaneous oximetry of 96%; lung scintigraphy revealed 12% of radiopharmaceutical activity outside the lungs, showing a small rightleft shunt, consistent with hepatopulmonary syndrome settlement.
Case 2
A 15-year-old girl was admitted to the Pediatric Hepatology outpatient clinic for elevated transaminases since the age of 8. Her family history revealed a 40-year-old mother with liver steatosis and a 41-year-old father with hypercholesterolemia and type 2 diabetes mellitus.
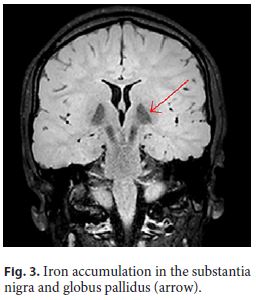
The patient had had neurological impairment since birth with global developmental delay, nonprogressive congenital ataxia, and oculomotor disturbances. By the age of 7, the first signs of a progressive kyphoscoliosis were noted, and, in the second decade of her life, her neurological condition evolved to a parkinsonian syndrome. Brain magnetic resonance imaging showed iron accumulation in the substantia nigra and globus pallidus (Fig. 3, arrow), consistent with neurodegeneration with brain iron accumulation diagnosis.
By the age of 3, she started losing adipose tissue in limb extremities and magnetic resonance imaging of the limbs supported the diagnosis of partial lipodystrophy. Molecular study of the AGPAT2 gene was normal. At the age of 14, autoimmune thyroiditis and thrombocytopenia were diagnosed. Raised transaminases were noted since the age of 8, with an asymptomatic raise to 10 times the upper limit of the normal range some weeks before admission to Hepatology consultation, at 15 years old.
Physical examination enhanced symmetrical distal absence of subcutaneous adipose tissue in upper and lower limbs, central obesity, kyphoscoliosis, ataxic gait, and upper limb tremor (Fig. 4, Fig. 5). There were no signs of chronic liver disease.
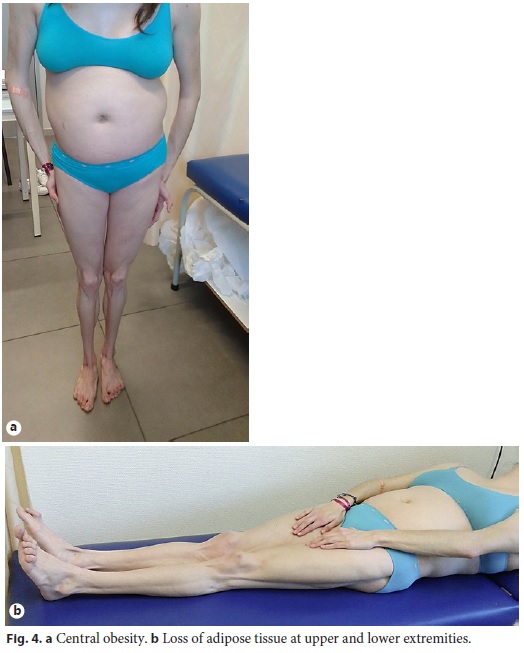
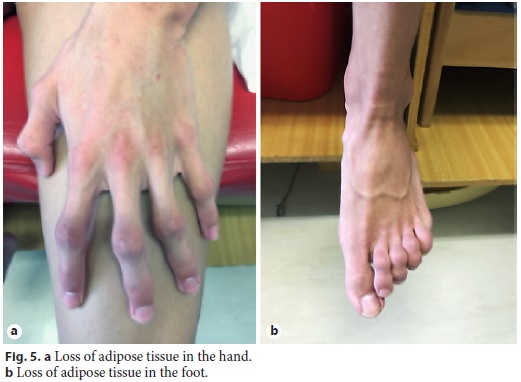
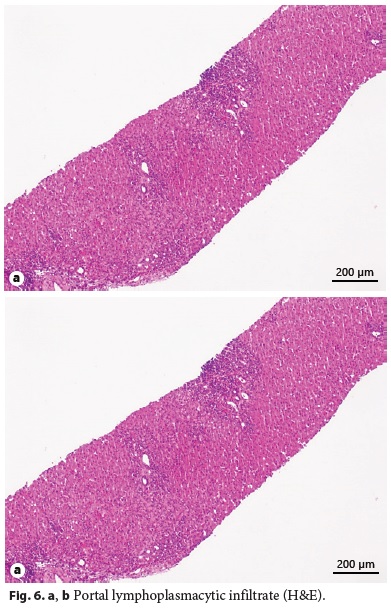
Laboratory tests evidenced elevated AST (461 IU/L), ALT (921 IU/L), GGT (93 IU/L), triglycerides (339 mg/dL), thrombocytopenia (63,000/μL), elevated IgG level (1,894 mg/dL), partial IgA deficiency (9.6 mg/dL), positive anti-LKM1 (1/640), and anti-LC1 antibodies. Leptin serum levels were normal. Abdominal ultrasound showed a liver with normal echogenicity and dimensions and a discrete homogeneous splenomegaly. Liver histology revealed a portal fibrous mass with partial nodular transformation, dense inflammatory infiltrate composed of lymphocytes and plasma cells, which crosses the limiting plate and invades the surrounding parenchyma (interface hepatitis). These findings were compatible with autoimmune hepatitis diagnosis (Fig. 6a, b), with a pretreatment score of 20 points [7].
The patient was treated with prednisolone (1 mg/kg/day) associated 4 weeks later with azathioprine (1.5 mg/kg/day), allowing progressive withdrawal of steroids. Complete normalization of transaminases, GGT, IgG levels, and platelet count were achieved within 2 months. Insulin resistance (HOMA-IR = 32.5) was noted and improved significantly under metformin therapy (500 mg b.i.d.). Currently, at the age of 16 years, she remains under treatment with azathioprine and prednisolone (5 mg/day), with no signs of recurrence of hepatitis, and she was classified as APL.
Discussion
The diagnosis of lipodystrophies relies on clinical history, physical examination, body composition, and metabolic status [1]. Clinical phenotype and immunological/ autoimmune abnormalities are important tools to distinguish genetic from acquired lipodystrophies [8]. Genetic testing can be helpful in the suspicion of familial lipodystrophies and should be performed in the presence of a suggestive family history[1] .
In case 1, lipodystrophy is generalized with predominant metabolic complications, such as insulin resistance, hypercholesterolemia, hypertriglyceridemia, ectopic fat accumulation manifested as a concentric left ventricular hypertrophy, and steatohepatitis that is progressing to cirrhosis. Since her family history and molecular study for congenital generalized lipodystrophy were negative, we assumed the diagnosis of AGL.
In case 2, despite the absence of an exhaustive genetic study, the lack of a family history and its association with autoimmune disorders (hepatitis, thyroiditis, and thrombocytopenia) favor the presumptive diagnosis of an acquired lipodystrophy. In this case, lipodystrophy is partial which can be easily misdiagnosed as common central obesity with metabolic syndrome. Liver involvement should have been suspected earlier, since the presence of hypertransaminasemia had been seen in a workup at the age of 8 years and not valorized and taken as incipient liver disease. Several neuromuscular abnormalities are described in lipodystrophies, particularly related to LMNA and BSCL2 gene mutation (genetic lipodystrophies)[9]. Neurodegeneration with brain iron accumulation, observed in this patient, is a neurologic disorder characterized by abnormal accumulation of iron in basal ganglia resulting in progressive dystonia, parkinsonism, and neuropsychiatric abnormalities [10] . Currently, there is no association described between this entity and lipodystrophy. Nevertheless, some studies suggest a relationship between serum leptin levels and Parkinsons disease [11].
The pathogenesis of fat loss in AGL remains unknown, despite several studies showing that 25% of cases are triggered by localized panniculitis with generalized progression, 25% have concomitant autoimmune disorders, and the remainder have idiopathic etiology [6, 8]. However, AGL is often associated with autoimmune diseases that are not the rule, as seen in case 1 [1, 8, 12]. APL pathogenesis is also unknown. As proposed for AGL, autoimmune theories have been proposed as an etiological cause for adipose tissue destruction due to its connection with autoimmune diseases, such as membranoproliferative glomerulonephritis, dermatomyositis, systemic lupus erythematosus, and autoimmune hepatitis [6, 8, 13]. Patient 2 has a clear autoimmunity reactivity.
Adipose tissue is a well-recognized active endocrine metabolic system [6]. Leptin is the leading key adipokine in metabolic homeostasis and neuroendocrine modulation with several functions, such as regulation of food intake, regulation of fatty acid metabolism, decrease in glucose synthesis, and increase in glucose uptake in muscle, lipid uptake in adipocytes, regulation of gonadal steroids secretion, and modulation of immune response [14–16]. Adipocytes provide a benign place to store lipids, protecting the organism from lipotoxicity [6]. Therefore, adipose tissue loss and resulting leptin deficiency can lead to serious clinical consequences developing at an early age, such as insulin resistance, impaired lipid storage with hypertriglyceridemia and fat ectopic accumulation in nonadipose tissue and impaired immune response with susceptibility for infection [8, 13].
Lipodystrophies are a secondary cause of fatty liver and by consequence one of the differential diagnosis of nonalcoholic fatty liver disease. Reduction of subcutaneous adipose tissue combined with leptin deficiency/resistance transforms the liver into the main organ of triglycerides accumulation [17]. Steatosis probably sensitizes the liver to metabolic injury leading to inflammation, necrosis, and culminating in fibrosis, as seen in the first case [17, 18]. Liver failure and hepatocellular carcinoma can also result from steatohepatitis progression; consequently, liver disease is an important cause of death in these patients. Histological assessment is imperative, allowing evaluation of steatohepatitis severity and the presence of autoimmune features, which can concomitantly be present in acquired lipodystrophies [6, 17]. Serum leptin levels are directly proportional to adipocytes tissue mass and are typically low in patients with generalized lipodystrophy (as seen in case 1), with variable values found in partial lipodystrophy [8, 19]. The severity of metabolic phenotype tends to be proportional to the grade of adipose tissue loss and, consequently, is tendentially worse in generalized lipodystrophy [20]. Metabolic complications are uncommon in APL, which is consistent with the less severe loss of adipose tissue and leptin levels [1, 6]. This was seen in case 2 in which the patient had a slight hypertriglyceridemia and insulin resistance compared with case 1.
Lipodystrophies have no cure and currently available therapies are used with the main goal of preventing or ameliorating associated comorbidities, such as metabolic disturbance [1, 8]. Energy-restricted diets have shown to improve metabolic complications but have to be implemented with caution in pediatric patients to avoid unbalanced requirements for an healthy development [1, 8]. In case 1, despite assertive pharmacological and nutritional management of metabolic complications, progressive worsening was observed with recent installation of hepatopulmonary syndrome. Physical activity was not an option due to arising cardiomyopathy. This is the natural history of generalized lipodystrophy, which is refractory to aggressive conventional therapy, as the main issue is the absence of leptin and its metabolic consequence [21]. In generalized lipodystrophies with severe leptin deficiency, the endocrine-metabolic disorder responsible for steatohepatitis with cirrhotic evolution may benefit from human recombinant leptin (metreleptin – Myalepta®, Aegerion Pharmaceuticals) treatment, as shown by several studies [8, 21]. Metreleptin has demonstrated to improve metabolic abnormalities with significant reduction in steatohepatitis activity scores, steatosis, serum triglycerides, transaminases, proteinuria, fasting glucose values, and hemoglobin A1c [17, 21]. Metreleptin is the only drug approved specifically for lipodystrophies and has recently been approved in the European Union as first-line therapy for endocrine-metabolic abnormalities treatment of generalized lipodystrophy as well as for hypoleptinemic partial lipodystrophy [1, 8, 22]. In case 2, leptin serum levels were normal, and no benefit would be expected from metreleptin treatment. Liver disease, in this case, was not fatty liver, but instead, an autoimmune hepatitis. Treatment was applied according to guidelines, so far with full success [23].
In conclusion, accurate diagnosis and effective management of lipodystrophy comorbidities are imperative to improve patient outcome, namely early recognition and adequate characterization of liver disease. Liver function monitoring and the performance of a timely liver biopsy is recommended.
References
1 Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, et al. The diagnosis and management of lipodystrophy syndromes: A multi-society practice guideline. J Clin Endocrinol Metab. 2016 Dec;101(12):4500–11. [ Links ]
2 Gupta N, Asi N, Farah W, Almasri J, Moreno Barrionuevo P, Alsawas M, et al. Clinical features and management of non-HIV related lipodystrophy in children: a systematic review. J Clin Endocrinol Metab. 2017 Feb 1;102(2):363–74. [ Links ]
3 Chiquette E, Oral EA, Garg A, Araújo-Vilar D, Dhankhar P. Estimating the prevalence of generalized and partial lipodystrophy: findings and challenges. Diabetes Metab Syndr Obes. 2017 Sep;10:375–83. [ Links ]
4 Commission of the European Communities. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions on Rare Diseases: Europes Challenges. 2016. [ Links ]
5 Adams C, Stears A, Savage D, Deaton C. Were stuck with what weve got: The impact of lipodystrophy on body image. J Clin Nurs. 2018 May;27(9-10):1958–68. [ Links ]
6 Nolis T. Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies. J Hum Genet. 2014 Jan;59(1):16–23. [ Links ]
7 Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999 Nov;31(5):929–38. [ Links ]
8 Cortés VA, Fernández-Galilea M. Lipodystrophies: adipose tissue disorders with severe metabolic implications. J Physiol Biochem. 2015 Sep;71(3):471–8. [ Links ]
9 Akinci G, Topaloglu H, Demir T, Danyeli AE, Talim B, Keskin FE, et al. Clinical spectra of neuromuscular manifestations in patients with lipodystrophy: A multicenter study. Neuromuscul Disord. 2017 Oct;27(10):923–30. [ Links ]
10 Gregory A, Hayflick SJ. Neurodegeneration with brain iron accumulation disorders overview. Gene Rev [Internet]. 2014;1–16. Available from: http://www.ncbi.nlm.nih.gov/books/NBK121988/ [ Links ]
11 Hsu RH, Lin WD, Chao MC, Hsiao HP, Wong SL, Chiu PC, et al. Congenital generalized lipodystrophy in Taiwan [Internet]. J Formos Med Assoc. 2018 Feb 22. pii: S09296646(17)30575-2.
12 Sahinoz M, Khairi S, Cuttitta A, Brady GF, Rupani A, Meral R, et al. Potential association of LMNA-associated generalized lipodystrophy with juvenile dermatomyositis. Clin Diabetes Endocrinol. 2018 Mar 27;4:6. [ Links ]
13 Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002 Oct;110(8):1093–103. [ Links ]
14 Kelesidis T, Kelesidis I, Chou S, Mantzoros CS. Narrative review: the role of leptin in human physiology: emerging clinical applications. Ann Intern Med. 2010 Jan 19;152(2):93–100. [ Links ]
15 Mechanick JI, Zhao S, Garvey WT. Leptin, an adipokine with central importance in the global obesity problem. Glob Heart. 2018 Jun;13(2):113–27. [ Links ]
16 Ceddia RB. Direct metabolic regulation in skeletal muscle and fat tissue by leptin: implications for glucose and fatty acids homeostasis. Int J Obes. 2005 Oct;29(10):1175–83. [ Links ]
17 Javor ED, Ghany MG, Cochran EK, Oral EA, DePaoli AM, Premkumar A, et al. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005 Apr;41(4):753–60. [ Links ]
18 Vajro P, Lenta S, Socha P, Dhawan A, McKiernan P, Baumann U, et al. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: position paper of the ESPGHAN Hepatology Committee. J Pediatr Gastroenterol Nutr. 2012 May;54(5):700–13. [ Links ]
19 Altay C, Seçil M, Demir T, Atik T, Akıncı G, Özdemir Kutbay N, et al. Determining residual adipose tissue characteristics with MRI in patients with various subtypes of lipodystrophy. Diagn Interv Radiol. 2017 Nov-Dec;23(6):428–34. [ Links ]
20 Berger S, Ceccarini G, Scabia G, Barone I, Pelosini C, Ferrari F, et al. Lipodystrophy and obesity are associated with decreased number of T cells with regulatory function and proinflammatory macrophage phenotype. Int J Obes. 2017 Nov;41(11):1676–84. [ Links ]
21 Brown RJ, Oral EA, Cochran E, Araújo-Vilar D, Savage DB, Long A, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. 2018 Jun;60(3):479–89. [ Links ]
22 European Medicines Agency. Myalepta (metreleptin): An overview of Myalepta and why it is authorised in the EU [Internet]. 2018. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/myalepta. [ Links ]
23 Mieli-Vergani G, Vervain D, Batman U, Czubkowski P, Debray D, Dezsofi A, et al. Diagnosis and management of pediatric autoimmune liver disease: ESPGHAN Hepatology Committee Position Statement. J Pediatr Gastroenterol Nutr. 2018 Feb;66(2):345–60. [ Links ]
Statement of Ethics
The authors have no ethical conflicts to disclose. Informed consent was obtained from the parents of both patients, authorizing publication of clinical data and photos with the guarantee of anonymity.
Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Sources
The authors have no funding source to declare.
* Corresponding author.
Ermelinda Santos Silva
Gastroenterology Unit, Pediatrics Division, Child and Adolescent Department
Centro Materno Infantil do Norte, Centro Hospitalar Universitário do Porto
Largo da Maternidade Júlio Dinis, PT–4050-651 Porto (Portugal)
E-Mail ermelinda.dca@chporto.min-saude.pt
Received: October 19, 2018; Accepted after revision: November 25, 2018
Acknowledgement
Many thanks to Prof. David Araújo-Vilar, Santiago de Compostela University, for his help with issues of diagnosis and management of case 1.
Author Contributions
Andreia Ribeiro has written the article and collected all the information from the other authors; José Ricardo Brandão provided all histological pictures and helped interpret them; Esmeralda Cleto followed one of the patients due to hematological disorders and helped interpret her management; Manuela Santos followed one of the patients due to Parkinson syndrome and provided MRI, as well as helped interpret the patients clinical condition; Teresa Borges followed both patients due to their endocrine-metabolic disorder and helped manage and interpret the lipodystrophy management; Ermelinda Santos Silva followed both patients due to their hepatic disease and was the person first responsible for these patients. All authors read the article and contributed to significative changes according to their specialization field.

