










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGE-Portuguese Journal of Gastroenterology
versão impressa ISSN 2341-4545
GE Port J Gastroenterol vol.24 no.5 Lisboa out. 2017
https://doi.org/10.1159/000461590
CLINICAL CASE STUDY
Uncertainties in the Management of a Lynch Syndrome Patient: A Case Report
Incertezas na Abordagem ao Doente com Síndrome de Lynch: A propósito de um Caso Clínico
Sara Camposa, Pedro Amaroa, Inês Cunhaa, João Fragab, Maria Augusta Ciprianob, Luís Toméa
aGastroenterology Department, and bPathology Department, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal
* Corresponding author.
ABSTRACT
Introduction: Lynch syndrome (LS), the most common hereditary colorectal cancer syndrome, is characterized by mutations in mismatch repair (MMR) genes leading to an increased cancer risk, namely colorectal cancer. Case: In the context of surveillance colonoscopy, a 40-mm flat lesion (0-IIa+b, Paris classification) was identified and submitted to piecemeal mucosal endoscopic resection in a 64-year-old LS patient with an MLH1 germline mutation (262delATC) and two previous segmental resections due to metachronous colorectal cancer. Pathology raised the suspicion of superficial submucosal invasive carcinoma with poor differentiation. Immunochemistry showed heterogeneous MLH1 expression and PMS2 loss. In a short-term follow-up colonoscopy, another 30-mm advanced carcinoma was identified. The patient was referred to surgery. Conclusion: This case raises several issues: (1) the potentially fast tumorigenesis and progression to carcinoma in LS and implications for endoscopic screening and surveillance; (2) pitfalls in the interpretation of MMR proteins immunochemistry; (3) the role of endoscopic resection in LS.
Keywords: Lynch syndrome; Immunochemistry; Endoscopic resection
RESUMO
Introdução: O síndrome de Lynch (SL), a causa mais frequente de cancro colorectal hereditário, é caracterizado por mutações nos genes de reparação do ADN e risco aumentado de cancro, nomeadamente colorectal. Caso: No contexto de vigilância endoscópica, uma lesão plana com 40mm (0-IIa+b, classificação de Paris) foi identificada e submetida a resseção endoscópica fragmentada num doente de 64 anos com SL com mutação germinativa do gene MLH1 (262delATC) e duas resseções segmentares prévias por cancro colorectal metacrónico. A histologia era suspeita de invasão submucosa superficial por carcinoma pouco diferenciado. A imunohistoquímica mostrou expressão heterogénea de MLH1 e perda de PMS2. O diagnóstico de um segundo carcinoma avançado com 30 mm em reavaliação endoscópica determinou a referenciação para cirurgia. Conclusões: Este caso levanta várias questões: (1) tumorigénese e progressão para carcinoma aceleradas no SL e implicações no rastreio e vigilância endoscópica; (2) problemas na interpretação da imunohistoquímica das proteínas MMR; (3) papel da resseção endoscópica no SL.
Palavras-Chave: Síndrome de Lynch; Imunohistoquímica; Resseção endoscópica
Introduction
Lynch syndrome (LS), the most frequent hereditary colorectal cancer (CRC) accounting for 1–3% of all CRC [1–5], is an autosomal dominant disorder in which there is a deleterious germline mutation in one of a set of DNA mismatch repair (MMR) genes – MLH1 , MSH2 , MSH6 , or PMS2 – or loss of expression of the MSH2 gene due to deletion in the EPCAM gene [6–9], with increased risk of cancer, namely CRC [1–5].
CRC in LS, comparing to sporadic CRC, is diagnosed in younger ages, more frequently localized in the right colon, with a faster sequence adenoma-carcinoma, distinct CRC histological characteristics, and a better prognosis [10].
Clinical Case
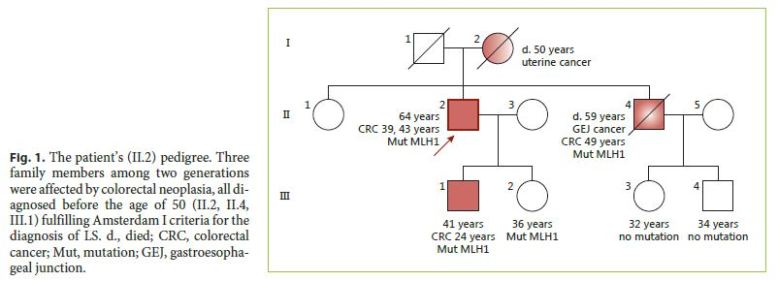
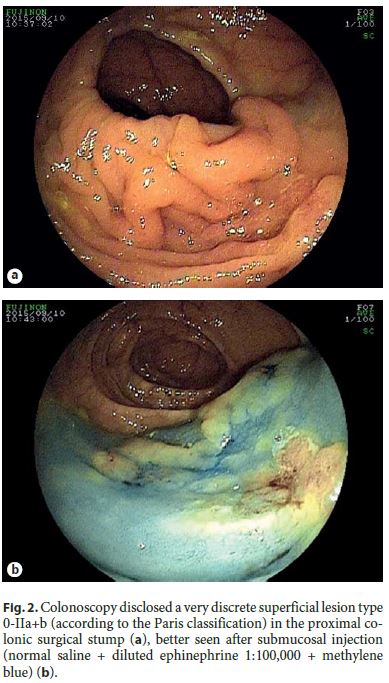
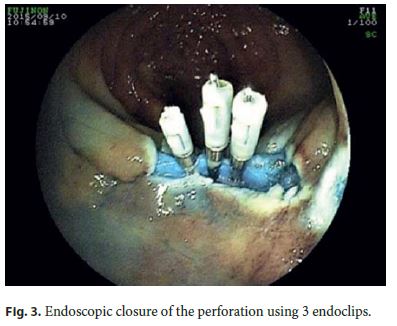
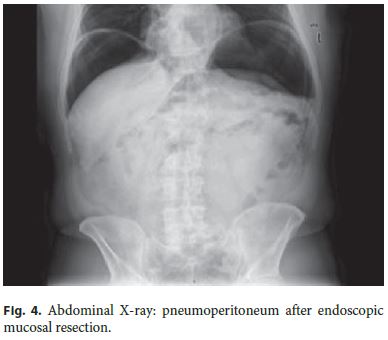
The authors describe a case of a 64-year-old male patient followed in Gastroenterology for 11 years due to LS. He had been previously submitted to colorectal surgery when he was 39 years old due to malignancy of the rectosigmoid junction. At the age of 43, he underwent a new surgery due to a metachronous malignant lesion in the ascending colon. Both lesions were treated while he was abroad, with no additional information available, namely the histopathologic features. LS diagnosis has been confirmed through genetic testing (mutation in codon 262 of MLH1 gene, 262del-ATC). Concerning his family history, he had a brother with surgically treated colon cancer at 49 years who died at 59 years due to gastroesophageal junction adenocarcinoma, a son with colon cancer at 24 years treated with curative surgery, and a daughter of 36 years without bowel lesions; all these relatives have a positive genetic testing for the culprit mutation and those still alive are under endoscopic surveillance. The mother died of uterine cancer when she was 50 years old (Fig. 1). The index patient started follow-up in our department at the age of 53 years. Since then, he had surveillance colonoscopies without sedation every 1–2 years, showing a right hemicolectomy with ileocolic anastomosis and a colorectal anastomosis at 10 cm of the anal verge. A few low-grade tubular adenomas, the largest with 12 mm, have been excised by polypectomy. In the last of these procedures, a superficial nonpolypoid 40-mm flat lesion (type 0-IIa+b, Paris classification) was identified in the proximal colonic surgical stump (Fig. 2). In the two previous endoscopic examinations, suboptimal bowel preparation (poor bowel preparation was found above the splenic flexure, Boston subscore 1) was noticed, and a shorter surveillance interval (yearly) and an optimized preparation protocol were proposed. A piecemeal endoscopic mucosal resection (EMR) was performed and complicated by a 6–7 mm perforation that was successfully closed with 3 long (clip arm length 9 mm) endoclips (EZClip HX-610-090L, Olympus TM ; Fig. 3). EMR was finished and there were no signs of residual lesion. During this colonoscopy, additional flat/sessile lesions with smaller size were identified throughout the remaining colon, four of which, with 5 to 12 mm, were also excised. However, considering the long duration of the procedure, the perforation (even if endoscopically treated), and patient discomfort probably related to pneumoperitoneum (no CO 2 insufflation available) (Fig. 4), other few similarly small and apparently unremarkable lesions were intentionally left behind. Inpatient conservative treatment with antibiotics and analgesia was proposed and the patient was discharged 5 days later with no further complications. All piecemeal EMR specimen fragments were retrieved and showed intraepithelial flat tubular and villous adenoma with high-grade neoplasia/intramucosal carcinoma with various patterns (tubular, solid, syncytial, signet ring, and mucinous). One of those fragments exhibited a solid and syncytial pattern associated with chronic inflammatory cells suspicious of focal superficial submucosal invasive carcinoma (possible sm1) with no lymphovascular invasion or tumor budding; the pathology was evaluated by two pathologists in our institution and further revised in a second institution. Immunohistochemistry (IHC) showed PSM2 loss of expression, and MLH1 heterogeneous expression pattern, negative in solid areas and positive in villous lesions; MSH6 and MSH2 were conserved (Fig. 5). CK7 and CK20 were negative. The larger (12 mm) of the remaining four excised lesions was a noninvasive neoplasia that also displayed a similar solid and syncytial pattern with intense chronic inflammatory infiltrate, while the others showed tubular adenoma with low- and high-grade dysplasia.
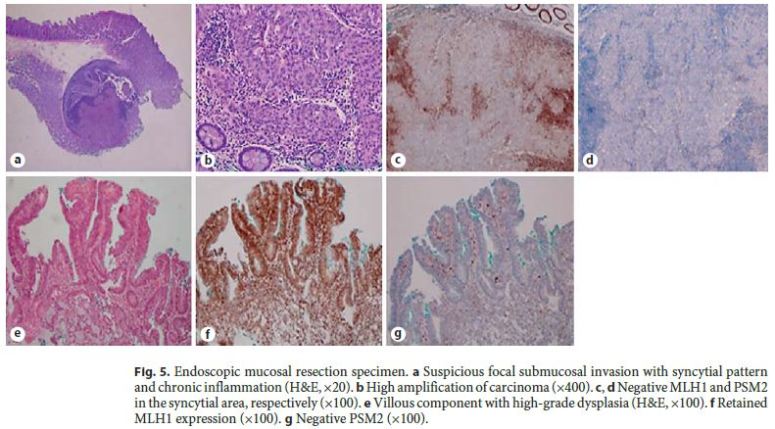
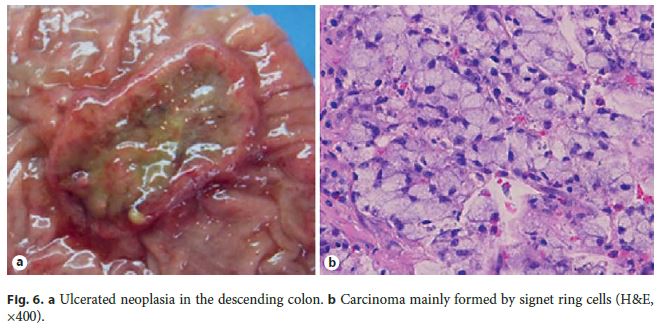
A multidisciplinary discussion on further management was carried out and additional surgery was considered. However, the patient preferred to be submitted to further endoscopic and imaging evaluation. Thoracic abdominal CT scan showed several millimetric pulmonary lesions, but FDG-F18-PET-CT scan confirmed no distant metastasis. A revision colonoscopy took place only 5 months later, showing a regular scar with absence of neoplastic residual tissue in the surgical colonic stump; however, an endoscopically advanced 30-mm ulcerated neoplastic lesion was identified 28 cm from the anal verge, corresponding to a carcinoma composed by cellular cords and signet ring cells within mucinous pools.
The patient was then referred to surgery, and coloprotectomy with definitive ileostomy was performed. Ileorectal anastomosis could not be accomplished due to severe rectal perianastomotic adherences and fibrosis.
Pathology showed an invasive carcinoma formed by irregular glands, a major component of signet ring cells (Fig. 6) and a component of extracellular mucin less than 50% of the neoplasia. The tumor invaded up to the muscularis propria without lymphovascular/neural invasion or metastatic disease in 21 lymph nodes (TNM classification: T2N0M0). There was no sign of residual lesion from the previous EMR in the surgical stump.
Discussion
LS diagnosis is based on clinicopathological features comprised in the Amsterdam (I or II) criteria or the revised Bethesda guidelines and should be confirmed at molecular level with genetic testing. Direct mutation screening, the approach followed in this case, is both time-consuming and expensive. Research has been done to define a more efficient workup algorithm, initially using tumor DNA microsatellite instability (MSI) and more recently tumor MMR protein detection by IHC to justify and direct genetic testing. The latter approach has gained preference as first line; however, MSI still has a role as an alternative in cases of inconclusive/normal IHC and a high clinical suspicion [11]. A missing protein suggests a mutation in the gene that codes for that protein. This is generally the case with loss of expression of MSH2 or MSH6; however, when MLH1 and PMS2 are lost, a BRAF mutation and/or MLH1 promoter hypermethylation, which may be involved in MSI-high CRC in older patients without LS, must be excluded before proceeding to MMR mutation testing.
In this case, tumor IHC was not mandatory because a diagnosis was already established by the identification of an MLH1 gene mutation; however, heterogeneous tumor MLH1 expression and PMS2 absence illustrates one pitfall of IHC that must be acknowledged. In their functional state, MMR proteins form heterodimers: MSH2 dimerizes with MSH6 and MLH1 is usually attached to PMS2 [11]. MLH1 germline mutations can be due to a nonsense mutation in two-thirds of the cases usually determining loss of MLH1 tissue expression; however, in the other one-third, a missense mutation results in an inactive mutant protein that is antigenically intact, producing a false-normal staining pattern in IHC [11]. In these cases, MLH1 antibodies are unable to detect all MLH1 abnormalities; the same may happen by an unknown mechanism even with protein-truncating mutations and large in-frame deletions in MLH1 [11]. Another possible explanation comes from the second hit that inactivates the second normal allele, which may also result in a nonfunctional antibody-binding MLH1 protein and variable staining patterns in IHC [11]. For these reasons, even though PMS2 mutations probably account for only 6% of all LS, the inclusion of PMS2 antibody in the IHC testing panel is mandatory because the absence of PMS2 expression may increase the accuracy by detecting up to 23% of MHL1-mutated tumors missed by MLH1-IHC [12].
LS management, when it concerns CRC prevention, is based on regular high-quality endoscopic surveillance [13, 14]. In this case, suboptimal bowel preparations may have hindered the detection of already existing lesions, namely the two larger ones.
Endoscopic treatment is the first-line approach of early lesions; total colectomy with ileorectal anastomosis is indicated for advanced neoplasia/lesions not removable by endoscopy. In this case, a 40-mm superficial neoplasia was completely removed by EMR, but it was not curative considering the poor differentiation and the suspected superficial submucosal invasion in the context of piecemeal resection [15]. Additional surgery was recommended in spite of the uncertainty if the criteria defining curative endoscopic resection, which were designed for sporadic neoplasia, should be similarly applied to LS. In fact, the prognosis of LS is more favorable for reasons still not clarified but eventually in relation to an intense immunological reaction, as was the case. Unfortunately, an endoscopically advanced neoplasia was detected in the post-EMR colonoscopy, making surgery mandatory. This lesion was missed earlier, probably due to the particular circumstances of the procedure (perforation, retrieval of all the fragments, long duration, patient discomfort).
The authors present herein a case of LS to draw attention to this syndrome with CRC predisposition and fast malignization of small nonpolypoid colonic lesions, where a specific protocol of colorectal surveillance is needed to ensure prevention against CRC. Additionally, the case underscores the peculiar histopathologic features of LS and the IHC pitfalls in cases with MLH1 mutations. Finally, the role of endoscopic resection in LS with large neoplastic lesions needs further evaluation and guidance.
References
1 Salovaara R, Loukola A, Kristo P, Kääriäinen H, Ahtola H, Eskelinen M, et al: Populationbased molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000;18:2193–2200. [ Links ]
2 Piñol V, Castells A, Andreu M, Castellví-Bel S, Alenda C, Llor X, et al: Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005;293:1986–1994. [ Links ]
3 Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomäki P, et al: Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998;338:1481–1487. [ Links ]
4 Barnetson RA, Tenesa A, Farrington SM, Nicholl ID, Cetnarskyj R, Porteous ME, et al: Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 2006;354:2751–2763. [ Links ]
5 Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al: Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851–1860. [ Links ]
6 Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, et al: The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027–1038. [ Links ]
7 Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, et al: Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993;75:1215–1225. [ Links ]
8 Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, et al: Mutation of a mutL homolog in hereditary colon cancer. Science 1994;263:1625–1629. [ Links ]
9 Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, et al: Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary nonpolyposis colon cancer. Nature 1994;368:258–261. [ Links ]
10 Giardiello FM, Allen JI, Axilbund JE, Boland CR, Burke CA, Burt RW, et al: Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-Society Task Force on colorectal cancer. Am J Gastroenterol 2014;109:1159–1179. [ Links ]
11 Shia J: Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. J Mol Diagn 2008;10:293–300. [ Links ]
12 de Jong AE, van Puijenbroek M, Hendriks Y, Topc C, Wijnen J, Auserns MG, Meijers-Heijboer H, Wagner A, van Os TA, Brocker-Vriends AH, Vasen HF, Morreau H: Microsatellite instability, immunohistochemistry and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res 2004;10:972–980. [ Links ]
13 Syngal S, Brand RE, Church JM, Giardello FM, Hampel HL, Burt RW: ACG Clinical Guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015;110:223–262. [ Links ]
14 Niv Y, Moeslein G, Vasen HFA, Karner-Hanusch J, Lubinsky J, Gasche C: Quality of colonoscopy in Lynch syndrome. Endosc Int Open 2014;2:E252–E255. [ Links ]
15 Pimentel-Nunes P, Dinis-Ribeiro M, Ponchon T, Repici A, Vieth M, De Ceflie A, et al: Endoscopic submucosal dissection: European Society of Gastrointestinal Endoscopy guideline. Endoscopy 2015;47:829–854. [ Links ]
Statement of Ethics
This study did not require informed consent nor review/approval by the appropriate ethics committee.
Disclosure Statement
The authors have no conflicts of interest to declare.
* Corresponding author.
Dr. Sara Campos
Gastroenterology Department, Centro Hospitalar e Universitário de Coimbra
Av. Bissaya Barreto – Praceta Prof. Mota Pinto
PT–3000-075 Coimbra (Portugal)
E-Mail saratcampos@gmail.com
Received: November 7, 2016; Accepted after revision: January 24, 2017
Acknowledgements
The authors would like to thank Prof. Dr. Fátima Carneiro, Pathology Department, Centro Hospitalar de S. João, for slides review and helpful discussion.

