










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Medicina Geral e Familiar
versão impressa ISSN 2182-5173
Rev Port Med Geral Fam vol.33 no.6 Lisboa dez. 2017
RELATOS DE CASO
Miastenia gravis, o diagnóstico no olhar: relato de um caso
Myasthenia gravis, the eye as diagnostic clue: a case report
Marisa Benigno Bizarro,* Marco André Oliveira,* Lurdes Nery,* Dora Isabel Alves*
*USF Grão Vasco, ACeS Dão Lafões
Endereço para correspondência | Dirección para correspondencia | Correspondence
RESUMO
Introdução: A miastenia gravis (MG) é uma doença neurológica autoimune crónica, que afeta as estruturas pós-sinápticas da junção neuromuscular dos músculos esqueléticos, caracterizada por fraqueza e fadiga muscular localizada ou generalizada.
Descrição de caso: Mulher de 31 anos, sem antecedentes pessoais de relevo. Recorreu à consulta da USF por dificuldade em encerrar de forma completa ambas as pálpebras e desconforto na região cervical anterior com início há um mês. Ao exame objetivo constatou-se aparente exoftalmia, tendo-se colocado inicialmente a hipótese de patologia tiroideia. Uma semana depois, para além das queixas iniciais, existia noção de voz nasalada e desconforto facial que agravava sobretudo ao final do dia, motivo pelo qual foi solicitada TC dos seios peri-nasais para exclusão de sinusite ou lesão ocupante de espaço. Na semana seguinte mantinha sintomatologia mas, após anamnese cuidada, constatou-se que apresentava também visão turva, parestesias faciais e disfagia. Perante esta sintomatologia foi excluída, em primeiro lugar, lesão cerebral ocupante de espaço por RMN-CE e suspeitou-se de patologia do foro neurológico e/ou muscular, nomeadamente MG, tendo sido referenciada para consulta de neurologia. A EMG com estimulação repetitiva e a pesquisa de anticorpos anti-AChR corroboraram a hipótese diagnóstica colocada. A resposta inicial à piridostigmina foi fruste, com impacto negativo na qualidade de vida da utente. Entretanto, após sucessivos reajustes terapêuticos, o seu estado clínico melhorou significativamente e neste momento encontra-se clinicamente estável com apenas limitações mínimas. Mantém-se em seguimento regular devido ao caráter crónico da doença e pelo risco de flutuações quer a nível da evolução clínica quer a nível da resposta ao tratamento.
Discussão: Este caso clínico constituiu um enorme desafio diagnóstico. Reflete a importância da anamnese, do índice de suspeição (decorrente do conhecimento teórico e da experiência clínica), da continuidade e articulação dos cuidados.
Palavras-chave: Miastenia gravis; Pálpebras; Fraqueza muscular; Diagnóstico; Relato de caso
ABSTRACT
Introduction: Myasthenia gravis (MG) is a chronic autoimmune neurologic disease, which affects the postsynaptic structures of the neuromuscular junction, characterized by localized or generalized weakness and muscle fatigue.
Case description: 31-year-old woman, without significant past medical history, reports difficulty in completely closing both eyelids and discomfort in the anterior cervical region, with onset one month ago. Physical examination revealed apparent exophthalmos, and the hypothesis of thyroid pathology was initially considered. A week later, in addition to the initial complaints, the patient also reported nasal voice and facial discomfort that worsened at the end of the day. A CT scan of the paranasal sinuses was requested to exclude the presence of sinusitis or space occupying lesion. The following week, the patient maintained the same symptoms but, after careful examination, it was verified that the patient also presented with blurred vision, facial paresthesia and dysphagia. The presence of an intracranial space occupying lesion was excluded by magnetic resonance imaging of the brain and then the hypothesis of a neuromuscular disease, namely MG, was considered. The patient was referred to a neurologist. The electromyography with repetitive nerve stimulation and the search for anti-AChR antibodies corroborated this diagnostic hypothesis. The initial response to pyridostigmine was poor, with a negative impact on the patients' quality of life. However, after successive adjustments, her clinical status has significantly improved and she is currently clinically stable with only minor limitations. Due to the chronic nature of the disease and the risk of fluctuations both in clinical evolution and response to treatment, the patient has regular follow-up appointments.
Discussion: This case report presented a great diagnostic challenge. It reflects the importance of anamnesis, high level of suspicion (due to theoretical knowledge and clinical experience), and of continuity and transitions of care.
Keywords: Myasthenia gravis; Eyelid; Muscle weakness; Diagnosis; Case report
Introdução
A miastenia gravis (MG) é uma doença neurológica autoimune das estruturas pós-sinápticas da junção neuromuscular dos músculos estriados esqueléticos, crónica, caracterizada por fraqueza e fadiga muscular localizada ou generalizada.1-2
Consiste numa patologia pouco comum, com uma prevalência mundial estimada em 20 por 100.000 pessoas. No entanto, estudos recentes afirmam que a sua prevalência tem aumentado nos últimos anos.3 Esta doença pode manifestar-se em todos os grupos etários e em ambos os géneros. Existe uma maior incidência no género feminino, entre a adolescência e os 40 anos3 e um aumento de novos diagnósticos em idades mais tardias.2
Relativamente à etiopatogenia, sabe-se que a presença de anticorpos contra recetores da acetilcolina (AChR) acarreta a destruição dos locais de ligação disponíveis para a acetilcolina na membrana pós-sináptica.2-3 Anticorpos contra outras proteínas da junção neuromuscular pós-sináptica, como a tirosina quinase músculo específica (MuSK), comprometem também a transmissão neuromuscular,2-3 que se irá manifestar como fraqueza músculo-esquelética. A etiologia para a síntese de anticorpos autoimunes permanece obscura. Em cerca de 50% dos doentes, a MG pode estar associada a hiperplasia folicular tímica e em 10% a timoma.4 Eventual associação com outras patologias autoimunes, especialmente tiroidite, lúpus eritematoso sistémico ou artrite reumatoide, tem sido constatada.5 Além disso, podem existir crises miasténicas induzidas por fármacos.
Habitualmente, a queixa inicial consiste em fraqueza muscular que envolve um grupo muscular específico em vez de fraqueza muscular generalizada.3 Frequentemente, os doentes notam que essa fraqueza muscular flutua de dia para dia, piorando com a atividade muscular continuada (fatigabilidade) e melhorando com o repouso e o frio.3 A fraqueza dos músculos oculares com ptose ou diplopia está inicialmente presente em 50% dos doentes e ocorre durante o curso da doença em 85-90%.6-7 A incapacidade em encerrar as pálpebras (fraqueza das pálpebras inferiores que expõe a esclera do olho),7 a mímica facial pobre e a dificuldade em soprar são sintomas de fraqueza dos músculos faciais.3 O envolvimento dos músculos bulbares é também comum, podendo condicionar disartria lingual, bucal ou palatal (voz nasalada), disfagia e dificuldades na mastigação.3 De referir que também pode estar presente proptose ligeira devida à fraqueza dos músculos extra-oculares.7
O curso da MG é variável, podendo variar desde MG ocular isolada a MG generalizada ou orofaríngea e, raramente, crises miasténicas com insuficiência respiratória com necessidade de ventilação mecânica.3,5 Cerca de 50 a 87% dos doentes têm doença generalizada nos primeiros 13-24 meses.6-7 Menos frequentemente, a doença pode permanecer exclusivamente nos músculos oculares.5
A abordagem diagnóstica assenta sobretudo no índice de suspeição (associado ao conhecimento teórico e experiência clínica), na eletromiografia (EMG) com prova de estimulação nervosa repetitiva e na pesquisa de autoanticorpos, em especial os anti-AChR.
Descrição do caso
Mulher de 31 anos, raça caucasiana, residente em Viseu e que trabalha como operária de loja. Insere-se numa díade nuclear altamente funcional, na fase I do Ciclo de Duvall e pertencente à classe média de Graffar. Sem antecedentes pessoais (patológicos ou cirúrgicos) relevantes. Sem hábitos medicamentosos (à exceção da contraceção oral hormonal combinada), tabágicos, alcoólicos ou consumo de drogas. Dos antecedentes familiares salienta-se mãe com história de carcinoma colorrectal, submetida a hemicolectomia em 2005, e pai com hipertensão e diabetes mellitus tipo 2.
A 28 de agosto de 2015 recorreu à consulta programada na USF por sensação de peso nos olhos, dificuldade em encerrar de forma completa ambas as pálpebras, com início há cerca de um mês. Referiu ainda desconforto na região cervical anterior, que não soube especificar. Negou outros sintomas, nomeadamente tremores, taquicardia, palpitações, sudorese intensa, ansiedade, irritabilidade ou perda de peso. Ao exame objetivo constatou-se encerramento incompleto das fendas palpebrais com aparente exoftalmia dos olhos. Apresentava pupilas isocóricas e isorreativas, movimentos oculares preservados e sem nistagmo, elevação mantida e simétrica das pálpebras superiores e sem assimetrias faciais. Na região cervical: sem tumefações visíveis, sem massas ou adenopatias palpáveis, tiroide mole de dimensões aparentemente normais, simétrica, móvel à deglutição e indolor à palpação. Orofaringe sem alterações. Perante estas queixas colocou-se a hipótese de se tratar de uma tiroidite pelo que foi solicitado doseamento de T4 livre, TSH, anticorpos antiperoxidase (ATPO), anticorpos antirecetor da TSH (TRAb) e ecografia da tiroide.
Uma semana depois, a 4 de setembro, recorreu à consulta com os resultados dos exames solicitados: apresentava TSH de 2,55 µmU/mL, T4 livre de 12,4 pmol/L (dentro dos parâmetros normais), ATPO e TRAb negativos e a ecografia da tiroide revelava glândula tiroideia de dimensões normais, homogénea, sem imagens nodulares aparentes. A utente apresentava persistência dos sintomas oculares, sem diminuição da acuidade visual ou diplopia. Referiu, ainda na consulta, um desconforto na face e garganta que não soube especificar e noção de voz nasalada, que agravava sobretudo ao final do dia. Ao exame objetivo apresentava voz nasalada, a palpação dos seios peri-nasais era indolor e o restante exame físico sobreponível ao anterior. Foi solicitada tomografia computadorizada (TC) dos seios peri-nasais para exclusão de sinusite ou de lesão ocupante de espaço.
A 10 de setembro regressa à consulta com o resultado da TC dos seios peri-nasais que não revelou alterações relevantes. Referiu que recorreu ao serviço de urgência do hospital de referência, onde foi observada por médico oftalmologista que recomendou colocação de penso ocular durante o sono e uso de lágrimas artificiais para proteção ocular durante o dia. Foi-lhe solicitada consulta de reavaliação após a realização dos exames solicitados nos cuidados de saúde primários. A utente mantinha dificuldade em fechar completamente os olhos, sobretudo o olho esquerdo (Figura 1A), e voz nasalada. Referiu ainda visão turva, parestesias faciais quando sorria, por vezes com dificuldade em sorrir e com dificuldade em engolir. Quando melhor questionada sobre este último sintoma, constatou-se que se tratava de disfagia cricofaríngea sobretudo para sólidos. Verificava agravamento dos sintomas oculares e faciais, sobretudo no final do dia. Negou défices de força muscular quer em repouso quer após o esforço físico, bem como desequilíbrio. Sem outros sintomas.
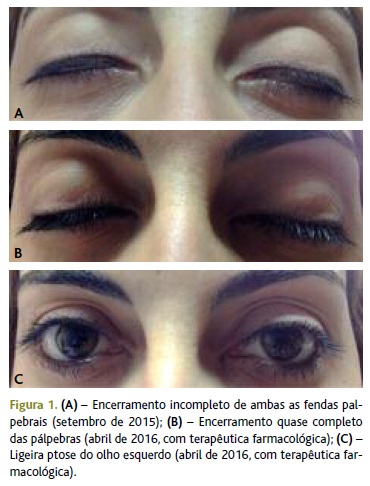
Perante uma doente com disfagia cricofaríngea e diminuição na força muscular para encerramento dos olhos seria pertinente, em primeiro lugar, excluir lesão cerebral ocupante de espaço e, na sua ausência, colocar a hipótese de patologia do foro neurológico e/ou muscular. Assim, realizou ressonância magnética crânio-encefálica (RMN-CE) que excluiu lesão ocupante de espaço, tendo sido referenciada, a 17 de setembro, como urgente para a consulta de neurologia do Centro Hospitalar Tondela-Viseu (CHTV, EPE) com a hipótese de diagnóstico de doença neuromuscular, nomeadamente miastenia gravis, para melhor avaliação e orientação terapêutica. Foi ainda aconselhada a colocação de frio/gelo para alívio das queixas oculares e manter as lágrimas artificiais, bem como o penso ocular durante o sono.
No decorrer das semanas seguintes iniciou disartria com agravamento progressivo. Apresentava discurso quase impercetível, o que condicionou significativamente o seu exercício profissional, motivo pelo qual consultou médico neurologista particular. Nesse contexto realizou EMG com estimulação nervosa repetitiva do nervo radial, que revelou resposta decremental significativa e reprodutível, compatível com a hipótese diagnóstica referida, tendo iniciado piridostigmina 60mg ½ + ¾ + ¾ + ¾ diariamente.
Na consulta hospitalar de neurologia, a 19 de outubro, apresentava poucas melhorias apesar da terapêutica instituída. O exame neurológico era sobreponível, exceto o surgimento de discreta ptose palpebral de predomínio esquerdo. Foi pedida TC do mediastino que excluiu timoma. O doseamento de anticorpos anti-AChR foi positivo, mas os anticorpos anti-Musk foram negativos. ANA, anticorpos anti-dsDNA e anti-mitocondriais M2 também positivos. Atendendo aos últimos resultados foi igualmente referenciada para a consulta de doenças autoimunes com a hipótese de possível coexistência de outros fenómenos autoimunes para além da MG.
Inicialmente a resposta à terapêutica foi fruste e, ao longo do seguimento hospitalar, houve necessidade sucessiva de ajustar a terapêutica para melhor controlo sintomático. Apesar do apoio que obteve por parte do cônjuge, família, amigos e do médico de família, houve um impacto negativo na sua qualidade de vida, que determinou o surgimento de sintomas como insónia inicial/terminal e humor deprimido.
A 20 de abril de 2016 apresentava-se clinicamente estável com limitações mínimas (Figura 1B e 1C), estando medicada com piridostigmina 60mg (dois comprimidos de 4/4 horas), prednisolona 20mg (um a dois comprimidos/dia em dias alternados), azatioprina 50mg 2 id, escitalopram 10mg id, esomeprazol 20mg id, carbonato de cálcio + colecalciferol 500mg/400 UI id e etinilestradiol + levonorgestrel 0,02/0,1mg. Aguardava estudo completo de doenças autoimunes e decisão clínica de eventual timectomia. Manteve seguimento regular em consulta com o seu médico de família e em consultas hospitalares de neurologia, medicina interna – doenças autoimunes e de oculoplastia.
Discussão
Este caso constituiu um enorme desafio diagnóstico. A sintomatologia inicialmente apresentada, nomeadamente a incapacidade de encerramento das pálpebras e a ligeira proptose, não fazem parte da clínica inicial típica da MG, mas a ptose palpebral com agravamento vespertino. Inicialmente dirigiu-se a investigação para patologias mais frequentes e só depois, durante o seguimento contínuo da doente e mediante uma anamnese cuidada com a exploração de outros sintomas que foram surgindo, foi possível chegar à hipótese de doença neuromuscular, como a MG. Apesar de se tratar de uma patologia rara, deve ser equacionada nos doentes que apresentem sintomas de fraqueza muscular ocular (ptose, diplopia), facial e bulbar (disfagia e disartria), agravando sobretudo ao fim do dia.
Além de um elevado índice de suspeição, para o diagnóstico de MG é requerido o doseamento de anticorpos anti-AChR2 devido à sua elevada especificidade. Estes anticorpos são positivos em 90% dos doentes com MG generalizada, mas apenas em 50-70% dos doentes com MG ocular.8 A pesquisa do anticorpo anti-Musk poderá ser positiva em mais de metade dos doentes com anticorpos anti-AChR negativos8 e, nestes casos, confirma o diagnóstico. A estimulação nervosa repetitiva (ENR) ou a eletromiografia de fibra única, caso a ENR seja duvidosa ou negativa, devem ser consideradas para auxiliar no diagnóstico uma vez que apresentam elevada especificidade.5,8 Na MG com sintomas limitados ao olho ou à face é essencial a exclusão de lesões intraorbitais ou intracranianas que possam comprimir os nervos cranianos, estando indicada a realização de TC ou RMN-CE.7 A TC do tórax deve ser realizada em todos os doentes recém-diagnosticados para exclusão de timoma ou hiperplasia tímica.7 Se existir dispneia, o teste de função pulmonar é mandatório.7
A estratégia terapêutica depende da gravidade da sintomatologia,9 podendo ser dividida, segundo a sua funcionalidade, em terapêutica sintomática, terapêutica modificadora de doença e intervenção terapêutica aguda.10 No tratamento sintomático utilizam-se os inibidores da acetilcolinesterase, como a piridostigmina, com o intuito de facilitar a transmissão neuromuscular.9-10 Relativamente à terapêutica modificadora de doença, os glucocorticoides e a azatioprina são os imunossupressores de primeira linha.10 Outros fármacos imunossupressores, como ciclosporina, metotrexato, micofenolato de mofetil e tacrolimus, podem ser utilizados no caso de contraindicação, intolerância ou controlo clínico inadequado com o tratamento de manutenção de primeira linha.9-10 Rituximab ou pulsos de ciclofosfamida podem ser usados como terapêutica de escalonamento do tratamento de manutenção. A timectomia pode ser também uma opção terapêutica, sobretudo na presença de timoma.10-11 O tratamento da crise miasténica ou a sua prevenção, em situações de MG instável, são realizados através de intervenções que reduzem os anticorpos circulantes como a plasmaferese, a imunoadsorção, a administração de imunoglobulina intravenosa ou com pulsos de metilprednisolona.10
Com um adequado acompanhamento, a maioria dos doentes mantém uma qualidade de vida satisfatória e uma esperança média de vida semelhante à população geral.11 Podem ocorrer exacerbações desencadeadas por eventos stressantes, nomeadamente infeções, exposição a fármacos, cirurgias, trauma, gravidez, neoplasias ou outros fatores. Apesar de a mortalidade ser rara, importa referir que as crises miasténicas com envolvimento dos músculos respiratórios podem ser potencialmente fatais se não reconhecidas e tratadas atempadamente.11
Os doentes devem ser ativos o quanto possível, devendo evitar atividade física sustentada e realizar repouso frequente.7 No caso de existir dificuldade no encerramento dos olhos, estes devem aplicar lágrimas artificiais ao longo do dia e penso ocular durante o sono. Na presença de disfagia podem utilizar espessante na dieta líquida com o intuito de prevenirem regurgitação nasal ou aspiração. Estes doentes necessitam, ainda, de capacitação para reconhecerem e reportarem de imediato uma crise respiratória eminente.
Além destes cuidados, o reconhecimento e o tratamento imediato de uma infeção são imprescindíveis, porque esta constitui um fator importante para o desenvolvimento de uma crise miasténica. Deve ser evitada, ainda, a prescrição de fármacos que possam desencadear uma exacerbação, nomeadamente alguns antibióticos (aminoglicosídeos, ciprofloxacina, clindamicina, eritromicina), corticoides em doses elevadas, propanolol, verapamil, fenitoína, entre outros.7
Em mulheres em idade fértil com MG, a decisão de engravidar deve ser tomada em conjunto com a equipa médica. Habitualmente estas mulheres não apresentam complicações graves na gravidez, sendo possível ocorrer MG transitória neonatal que geralmente reverte após três semanas.12 A terapêutica instituída pode ser prejudicial para o desenvolvimento fetal, mas existem fármacos que parecem ser seguros.12 Já o curso da MG durante a gravidez é imprevisível, podendo ocorrer agravamento, remissão ou estabilidade da sintomatologia.12 A gravidade dos sintomas, bem como a mortalidade materna, são maiores nos primeiros dois anos após o início da doença e mínimas sete anos após o seu diagnóstico, motivo pelo qual é aconselhável protelarem a gravidez pelo menos dois anos após o diagnóstico da patologia.12
Deste modo, estes doentes requerem uma articulação adequada entre os dois níveis de cuidados de saúde. Por um lado, devem manter a vigilância regular em consultas de neurologia devido ao caráter crónico da doença e pelo risco de flutuações, quer a nível da evolução clínica quer a nível da resposta ao tratamento. Por outro lado, necessitam de um seguimento nos cuidados de saúde primários, com o seu médico de família, que passa a ter um papel importante quer na capacitação para a doença e para os cuidados a ter, quer no acompanhamento próximo e contínuo, com especial atenção não só a sinais de exacerbações agudas ou aos seus fatores de risco, mas também ao estado emocional e à dinâmica familiar e, ainda, ao planeamento familiar em mulheres em idade fértil.
A descrição deste caso clínico, que retrata uma patologia rara mas importante nos cuidados de saúde primários, pretendeu relembrar a existência desta doença e sintetizar a sua abordagem diagnóstica e terapêutica. O médico de família desempenhou aqui um papel essencial pelo reconhecimento precoce da patologia, pelo acompanhamento contínuo e pela articulação dos cuidados.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Tsonis AI, Zisimopoulou P, Lazaridis K, Tzartos J, Matsigkou E, Zouvelou V, et al. MuSK autoantibodies in myasthenia gravis detected by cell based assay: a multinational study. J Neuroimmunol. 2015;284:10-7. [ Links ]
2. Binks S, Vincent A, Palace J. Myasthenia gravis: a clinical-immunological update. J Neurol. 2016;263(4):826-34. [ Links ]
3. Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8(5):475-90. [ Links ]
4. Willcox N, Leite MI, Kadota Y, Jones M, Meager A, Subrahmanyam P, et al. Autoimmunizing mechanisms in thymoma and thymus. Ann N Y Acad Sci. 2008;1132:163-73. [ Links ]
5. Sieb JP. Myasthenia gravis: an update for the clinician. Clin Exp Immunol. 2014;175(3):408-18. [ Links ]
6. Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve. 2008;37(2):141-9. [ Links ]
7. Jowkar A, Goldenberg WD, Shah AK. Myasthenia gravis. Medscape.com [homepage]. Medscape; 2017 [cited 2017 Dec 11]. Available from: https://emedicine.medscape.com/article/1171206-overview [ Links ]
8. Benatar M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul Disord. 2006;16(7):459-67. [ Links ]
9. Jones LA, Robertson NP. An update on treatments in myasthenia gravis. J Neurol. 2017;264(1):205-7. [ Links ]
10. Melzer N, Ruck T, Fuhr P, Gold R, Hohlfeld R, Marx A, et al. Clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurological Society. J Neurol. 2016;263(8):1473-94. [ Links ]
11. Gilhus NE, Nacu A, Andersen JB, Owe JF. Myasthenia gravis and risks for comorbidity. Eur J Neurol. 2015;22(1):17-23. [ Links ]
12. Chaudhry SA, Vignarajah B, Koren G. Myasthenia gravis during pregnancy. Can Fam Physician. 2012;58(12):1346-9. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Marisa Benigno Bizarro
E-mail: marisabizarro@gmail.com
Conflitos de interesse
Os autores declaram não ter quaisquer conflitos de interesse.
Recebido em 14-06-2016
Aceite para publicação em 11-12-2017

