










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Medicina Geral e Familiar
versão impressa ISSN 2182-5173
Rev Port Med Geral Fam vol.32 no.2 Lisboa abr. 2016
RELATOS DE CASOS
Apenas mais um acidente de trabalho? Relato de um caso clínico de coreia de Huntington
Just another work accident? A case report of Huntington chorea
Hugo Taveira Cunha,* Filipa Borges Lopes*
*Médicos Internos de Medicina Geral e Familiar. USF Arquis Nova, ULS Alto-Minho
Endereço para correspondência | Dirección para correspondencia | Correspondence
RESUMO
Introdução: As patologias que cursam com movimentos involuntários anormais, também designadas por discinésias, podem manifestar-se como coreia, atetose, tremor, mioclonia, asterixis, espasticidade, distonia ou tiques. A doença de Huntington é a causa mais comum de coreia hereditária, sendo causa de morbilidade importante e morte precoce. Os conviventes mais próximos são frequentemente os primeiros a notar as alterações coreicas. O médico de família, pelo acompanhamento dos seus utentes, ocupa uma posição privilegiada na identificação destes distúrbios do movimento.
Descrição do caso: Utente do sexo masculino, 41 anos de idade, raça caucasiana, casado. Desempregado (operário da construção civil até há um ano). Inserido numa família nuclear. Sem antecedentes pessoais de relevo. História familiar de pai falecido aos 56 anos com Parkinson, segundo descrição dos familiares. Dois filhos gémeos (5 anos de idade), saudáveis até à data.
Um familiar recorreu ao médico de família em janeiro de 2014, referindo que o utente apresentava quadro com três anos de evolução de “movimentos descoordenados e desajeitados” (sic) dos membros, alterações de memória e disartria. Essas alterações seriam semelhantes às que o pai do utente apresentava. Concomitantemente destacou a história de acidentes laborais na construção civil, o mais grave em 2012 (corte na mão direita com rebarbadora, com sequelas funcionais definitivas). Confrontado com a preocupação dos familiares, o utente recorre ao seu médico de família um mês mais tarde. Foi referenciado à consulta de neurologia, onde se diagnosticou doença de Huntington como causa do quadro de coreia.
Comentário: A história clínica (onde podem ser relevantes os sinais notados por familiares/conviventes) e o exame físico (com alterações típicas no exame neurológico) são o principal método de diagnóstico das doenças do movimento. As alterações motoras da coreia incluem incapacidade de manutenção de um movimento numa dada tarefa, podendo, como neste caso, predispor para acidentes.
Palavras-chave: Acidente Ocupacional; Discinésia; Doença de Huntington; Família; Médico de Família.
ABSTRACT
Introduction: Abnormal involuntary movements, also known as dyskinesias, may manifest as chorea, athetosis, tremor, myoclonus, asterixis, spasticity, dystonia and tics. Huntington disease is the most common cause of hereditary chorea and is a cause of morbidity and premature death. Relatives and others living with the patient are often the first to notice chorea in a patient. Family physicians have a special opportunity to diagnose movement disorders.
Case description: We present the case of a 41 year-old married Caucasian male living in a nuclear family. He was a construction worker but had been unemployed for one year. There was no relevant previous medical history. His father died at age 56 with “Parkinson disease”, according to one family member. He was the father of two healthy 5 year-old twin boys.
A relative of the patient went to the family physician in January 2014, stating that the health of the patient had changed in the past three years. He noted ‘uncoordinated and clumsy movements’ of the limbs, memory changes, and speech difficulties. These changes were similar to the abnormal movements that the patient’s father had shown. The relative also highlighted the history of a work accident he suffered in construction work in 2012, in which he a cut his right hand on a grinding wheel, with permanent loss of function. Due to the concerns of the family, the patient visited the family physician one month later. He was referred for consultation with a neurologist and Huntington disease was diagnosed as the cause of the chorea.
Comment: Movement disorders are diagnosed by taking a good clinical history (including obtaining information from family members and others living with the patient) and careful physical examination. Chorea may include the inability to maintain movement during motor tasks and this may predispose to accidents, as in this case.
Keywords: Dyskinesia; Family; Family Physician; Huntington Disease; Occupational Accident.
Introdução
As patologias que cursam com movimentos involuntários anormais são clínica e patologicamente heterogéneas. Manifestam-se clinicamente por alterações do movimento como tremor, coreia e/ou atetose, mioclonia e/ou asterixis, espasticidade, distonia ou tiques.1-2 A avaliação meramente clínica de cada caso permitirá classificá-lo em cada um desses grupos de alterações do movimento, que correspondem a grupos de patologias neurológicas do movimento com a mesma designação. Uma vez feita essa classificação diagnóstica, apresentam-se vários diagnósticos diferenciais como causa específica da alteração do movimento determinada clinicamente. Essa investigação é geralmente do âmbito e da competência da especialidade de neurologia, podendo requerer meios auxiliares de diagnóstico específicos.
Um destes grupos de doenças é a coreia. Coreia (palavra que deriva do latim de “dança”) define-se pelo quadro clínico que cursa com movimentos involuntários espontâneos, rápidos e abruptos, sem padrão de previsibilidade no tempo e na distribuição corporal.1-3 Estes movimentos dão a aparência ao comum observador de uma “postura inquieta”. A “impersistência motora” é um achado típico no exame físico neurológico.
Não está actualmente bem definida a prevalência da coreia na população geral. Sabe-se que a coreia, ou doença de Huntington, é a causa hereditária mais frequente de coreia, com uma prevalência mundial de 3 por 100.000 habitantes.4-6 Contudo, as etiologias secundárias são as causas mais frequentes da coreia no seu global.6-7 Nestas incluem-se causas iatrogénicas (e.g., o tratamento com levodopa em doentes com doença de Parkinson ou o tratamento com neurolépticos em doentes psiquiátricos), reumáticas (coreia de Sydenham e coreia da gravidez) ou por doenças sistémicas, ocorrendo, por exemplo, no lúpus eritematoso sistémico, tirotoxicose, no contexto de uma síndroma paraneoplásica ou na infecção VIH-SIDA.6,8-9
A doença de Huntington é uma doença monogénica causada por uma mutação no gene da proteína huntingtina, tendo um padrão de transmissão hereditária autossómico dominante.4-5 Os indivíduos afectados tipicamente possuem pelo menos um alelo desse gene com 40 ou mais repetições de sequências CAG.6,10 Nos portadores dessas alterações verifica-se penetrância completa da doença, enquanto nos portadores do gene com 36 a 39 repetições CAG se verifica penetrância incompleta da doença. Regra geral, quanto maior é o número de repetições CAG, mais precocemente se iniciam as primeiras manifestações da doença nos indivíduos portadores da mutação e mais rápida será a progressão clínica da doença desde que se inicia.6,10 Os primeiros sintomas ocorrem tipicamente entre os 30 e os 50 anos de idade. A progressão da doença é inexorável, apesar da rapidez ser variável.6,8 Não tem cura, resultando numa diminuição da esperança de vida dos indivíduos afectados. As primeiras manifestações clínicas ocorrem tipicamente entre os 30 e os 50 anos de idade, com morte geralmente ao fim de 10 a 25 anos.6,11-14 A doença progride com deterioração motora, com agravamento lento e progressivo da coreia, culminando em imobilidade geral e fraqueza dos músculos da deglutição e da respiração em estádios avançados.6,8 Além das alterações motoras que dominam o quadro desde o início, mais tardiamente dá-se a deterioração cognitiva, com instalação de um quadro demencial que afecta principalmente as funções executivas e surgem frequentemente alterações do humor (e.g., depressão) e/ou do comportamento (como agitação e agressividade).6,8 Os tratamentos disponíveis visam a melhoria da qualidade de vida dos doentes, mediante o controlo dos sintomas motores (com fármacos neurolépticos), sintomas psiquiátricos (fármacos antidepressivos e/ou antipsicóticos) e deterioração cognitiva (estimulação mediante fisioterapia, terapia ocupacional, etc.), não existindo actualmente tratamentos específicos dirigidos à doença que alterem o seu curso.7-8 A morte não advém directamente da doença, mas das comorbilidades a ela associadas. As grandes causas de morte dos indivíduos afectados pela doença são a pneumonia (que, em parte, estará relacionada com a deterioração da função de deglutição e respiração, com aspiração consequente) e eventos cardiovasculares.15
O diagnóstico da doença de Huntington baseia-se nos sinais e sintomas de coreia num indivíduo com história familiar confirmada de doença. Actualmente, a possibilidade de pesquisa da alteração genética no gene da proteína huntingtina permite confirmar analiticamente a doença. O estabelecimento do diagnóstico definitivo é feito na especialidade de neurologia, após exclusão de causas secundárias (atrás referidas), achados imagiológicos sugestivos e genotipagem.6,8 O estudo genético possibilita também um melhor aconselhamento genético, quer a indivíduos portadores/doentes em fase pré-concepcional quer a indivíduos em risco de serem portadores. Actualmente, é possível prevenir a transmissão da doença para a descendência, quer através do diagnóstico pré-natal quer por diagnóstico genético pré-implantação de embriões, durante a reprodução medicamente assistida.8
A clínica constitui a base do diagnóstico das patologias que cursam com movimentos involuntários anormais. Na neurologia, as doenças neurológicas do movimento, como grande grupo patológico, são frequentes.2 O presente caso destaca a abrangência de actuação do médico de família como central para a detecção de patologia específica, neste caso neurológica. O seguimento do utente ao longo da sua vida e o conhecimento e consideração do seu meio familiar e social constituem ferramentas diagnósticas.
Descrição do caso
O caso reporta-se a um homem de 41 anos de idade de raça caucasiana, armador de ferro da construção civil. Tinha como habilitações literárias o 6.o ano de escolaridade e pertencia a uma classe social média, segundo a classificação de Graffar. Era casado e estava inserido numa família nuclear na fase III do Ciclo de Vida de Duvall.
Tratava-se de um utente sem antecedentes pessoais médicos ou cirúrgicos de relevo e sem medicação habitual. Relativamente aos hábitos de consumo era fumador de cerca de 20 cigarros por dia, desde há 20 anos (20 unidades maço/ano), sem qualquer consumo habitual de bebidas alcoólicas. Dos antecedentes familiares destacava-se o pai, falecido aos 56 anos de idade, com condição descrita pelos familiares como Parkinson, que se teria iniciado por volta dos 45 anos. A mãe tinha falecido aos 36 anos de idade com cancro da mama. Quanto à descendência, tinha dois filhos de sexo masculino, com cinco anos de idade (gémeos heterozigóticos), saudáveis até à data. Não havia conhecimento sobre patologias dos avós e das circunstâncias/causas de morte. O tio sofria de hipertensão arterial, sem outras patologias conhecidas e as suas primas eram saudáveis até à data (genograma na Figura 1).
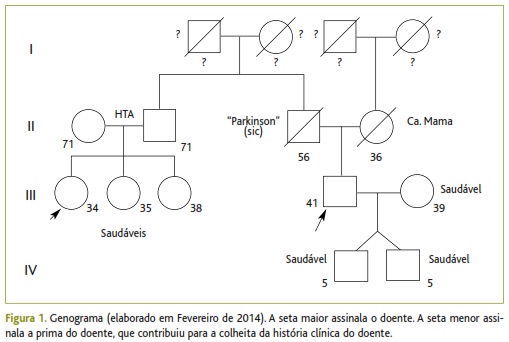
Tratava-se de um utente saudável até ao ano de 2010, sem qualquer intercorrência de saúde. Nesse ano iniciou história de vários acidentes de trabalho minor, provocados pelo manuseio de ferramentas e máquinas de trabalho da construção civil, algumas delas de corte de cimento ou ferro, que resultaram em traumatismos dos membros superiores ou inferiores. Na maioria das vezes (cerca de quatro episódios no total, ocorridos até 2012), os traumatismos não levaram o doente a recorrer a qualquer serviço de saúde, tratando-se por si mesmo no domicílio, sem resultarem daí quaisquer sequelas funcionais.
Em agosto do ano de 2012, o utente sofre um acidente no seu trabalho enquanto manuseava uma máquina de corte de ferro (rebarbadora). Desse acidente resultou traumatismo cortante da mão direita, com secção dos tendões flexores do 4.o e 5.o dedos e do nervo cubital. Foi assistido pelos serviços médicos da seguradora de trabalho, da empresa em que trabalhava, durante cerca de um ano até ter recebido alta médica. Durante esse período foi submetido a duas intervenções cirúrgicas à respectiva mão, com tentativa de reparação osteotendinosa da lesão e posterior intervenção de enxerto do nervo cubital. Ao fim de quase um ano de fisioterapia, teve alta com sequelas funcionais irremediáveis, nomeadamente com limitação da flexão e força preênsil dos 4.o e 5.o dedos, com atrofia dos músculos próprios da mão associada (condicionada pela lesão nervosa).
Cerca de três meses após o utente ter recebido alta médica pelos serviços da seguradora do trabalho, uma prima do utente recorre sozinha ao médico de família, que era comum aos dois utentes. Recorre à consulta pela preocupação crescente em relação a alterações que notava no seu familiar, que seriam progressivamente mais notórias desde há cerca de três anos. Segundo ela, o utente vinha desenvolvendo desde essa data alterações que a própria descrevia como sendo “movimentos desajeitados e descoordenados” dos membros, comparando-os a “trémulo” e que aparentemente também perturbavam por vezes a marcha (“desajeitada”). Nunca terão ocorrido quedas ou desequilíbrio associados. Referia ainda disartria aparente, ligeira, e esquecimento/falta de memória, ocasionalmente notória. Na sua perspectiva, o quadro descrito poderia ser causa dos acidentes laborais nos últimos três a quatro anos, ambos os factos coincidentes no tempo. A prima do utente referiu ainda que as alterações eram idênticas às verificadas no pai do utente, que já tinha falecido. Eram alterações que terão sempre sido assumidas e descritas por toda a família como Parkinson, sem no entanto ser claro se teria sido alguma vez avaliado e diagnosticado medicamente. Seria com essas alterações, com evolução desfavorável progressiva, que teria morrido. As alterações notadas e a hipotética relação de causalidade estabelecida eram, para a prima, fonte de preocupação e ansiedade. Desta forma, esse foi o motivo da consulta, tentando um pedido de auxílio ao médico de família comum a ambos. De referir que o utente não reconhecia os sinais notados pelos familiares, negando, além disso, qualquer sintoma. Além disso, tratava-se de um utente muito pouco utilizador dos serviços da sua USF e do seu médico de família, sendo saudável até à data do início das alterações clínicas. Nessa consulta foi esclarecido à prima o direito que só o utente tem de decidir acerca da sua própria saúde.
Cerca de um mês depois da referida consulta com a prima do utente, o próprio doente teve consulta programada com o médico de família, marcada por sua iniciativa após ter sido confrontado mais directamente pelos familiares, que lhe revelaram medo e preocupação pelas alterações que vinha notando em si. Sozinho na consulta, o doente contou uma história clínica semelhante à da prima. O quadro clínico consistiria desta forma em “movimentos desajeitados e descoordenados” dos membros inferiores e superiores, de predomínio superior e à esquerda. Eram mais notórios em situações em que desempenhava uma determinada tarefa motora, por comparação a quando se encontrava em repouso (predomínio de “intenção”), tendo a sensação de agravamento da intensidade em situações de nervosismo ou ansiedade. Ainda que de menor intensidade, comparou qualitativamente os seus movimentos ao “trémulo” que o pai desenvolveu nos últimos anos de vida. Esse quadro sempre terá sido assumido pela família como Parkinson, não sendo possível, no entanto, saber se a situação terá sido devidamente estudada e diagnosticada. Além disso, o doente assumia pequenas alterações perturbadoras da marcha, “desajeitada”, sem nunca ter desequilíbrio ou história associada de quedas. Admitiu que as alterações pudessem estar presentes nos últimos anos sem, no entanto, conseguir ser tão preciso como a prima (segundo a qual teria iniciado e evoluído progressivamente desde há três ou quatro anos), mas afirmando que eram mais notórias e mantidas durante o último ano. No exame físico estavam presentes alterações neurológicas, nomeadamente disartria ligeira, movimentos coreicos dos membros superiores e da cabeça, alterações dos membros inferiores notórias na marcha (marcha “coreica”) e impersistência motora notória na protrusão sustentada da língua. Não apresentava qualquer outra alteração do exame neurológico, como alterações da força muscular ou da sensibilidade dos membros, alterações nos testes dos pares cranianos (incluindo alterações da mímica facial ou bradicinesia facial aparente), alterações dos reflexos osteotendinosos rotulianos, bicipitais ou estilo-radiais, ou dismetrias nas provas dedo-nariz e calcanhar-joelho. Além disso, foi verificada negatividade para a prova de Romberg e Mingazzini. Não se verificaram tremores de repouso, de intenção ou posturais, nem fenómenos de rigidez muscular (de espasticidade muscular ou fenómeno de “roda-dentada” na mobilização passiva do cotovelo).
Perante a avaliação clínica compatível com coreia, o doente foi referenciado ao serviço de neurologia do hospital de referência da sua zona geográfica. O doente teve consulta de neurologia dois meses depois, onde foi confirmada coreia, de início insidioso. Atendendo à idade do doente, ausência de outra patologia conhecida e de medicação habitual, distribuição corporal dos achados físicos e história familiar sugestiva, foi estabelecido clinicamente o diagnóstico de doença de Huntington. O estudo etiológico da coreia incluiu exames complementares de diagnóstico, como estudo analítico (sem alterações do hemograma, velocidade de sedimentação, função renal, ionograma, perfil hepático, função tiroideia, proteínas totais e electroforese de proteínas, marcadores infecciosos ou de marcadores de auto-imunidade) e ressonância magnética (onde se notou diminuição dos núcleos caudados). Os resultados foram compatíveis com coreia de Huntington, excluindo causas secundárias de coreia. Além disso, foram avaliadas funções cognitivas recorrendo ao Exame Breve do Estado Mental (ou Mini-Mental State Examination), com um resultado no limite inferior da normalidade (resultado de 26, numa pontuação máxima possível de 30).
Actualmente, o doente é seguido na consulta de neurologia, estando medicado sintomaticamente com haloperidol na dose de 1mg por dia. Realizou estudo genético para a mutação da doença de Huntington, cujo resultado ainda não se encontra disponível.
Comentário
Neste caso clínico, como habitualmente descrito para as doenças que cursam com movimentos involuntários anormais (ou discinésias), a história clínica e o exame físico foram os pilares da sua detecção e avaliação. Estes permitiram a classificação das alterações do movimento, permitindo um diagnóstico final específico.
As alterações clínicas típicas das coreias terão estado provavelmente associadas à predisposição para a ocorrência dos acidentes de trabalho. Essa relação é apoiada pela sobreposição temporal da história de acidentes laborais e da história de alterações clínicas do movimento. Os acidentes poderão até ter constituído os primeiros sinais da alteração do estado de saúde do utente, numa análise a posteriori.
As alterações motoras da coreia incluem incapacidade de manutenção de um movimento numa dada tarefa, a chamada impersistência motora. Este é um sinal que pode ser pesquisado no exame físico pelos testes da protrusão da língua, no qual haverá tipicamente uma protrusão brusca da língua quando isso é solicitado ao doente. O doente é incapaz de manter a língua protraída de forma constante, movendo-a involuntariamente. Essa impersistência motora pode também ser evidente na incapacidade de manter a pressão de aperto quando o doente é solicitado a apertar a mão do examinador. Desta forma, esta alteração motora típica é compatível com o comprometimento da preensão e execução das tarefas motoras durante a actividade laboral - neste caso, enquanto o doente manuseava e segurava em máquinas de corte de ferro e cimento. Sendo máquinas já de si com elevado grau de risco para o seu utilizador, as alterações clínicas terão predisposto o doente para os acidentes ocorridos.
Apesar de o médico de família fazer o seguimento longitudinal no tempo dos seus utentes, neste caso o reconhecimento do padrão anormalmente mais frequente de acidentes de trabalho foi inicialmente dificultado. Por um lado, pelo facto de se tratar de um utente pouco utilizador dos serviços de saúde prestados na sua USF, compatível com a sua faixa etária (jovem adulto) e antecedentes pessoais de saúde (saudável até aí). Por outro lado, só tinha sido até aí dada maior relevância ao último dos acidentes de trabalho (em 2012), com sequelas graves, tanto pelo próprio doente como pelo médico de família.
Na literatura está descrito que os primeiros sinais das doenças que cursam com movimentos involuntários são frequentemente notados pelos familiares/conviventes mais próximos, antes ainda do próprio doente.16 Neste caso clínico ficou patente a importância do meio familiar envolvente do doente, sendo esses os primeiros a notar os primeiros sinais da doença. Além disso, ainda relacionaram a progressão da doença com os acidentes de trabalho ocorridos, suspeitando de uma relação causal. Relacionaram também as alterações clínicas com as verificadas no pai do doente, suspeitando de um padrão hereditário. O medo de uma doença por detrás dessas alterações notadas no ente querido terá sido fonte de preocupação e ansiedade, resultando na sinalização e pedido de auxílio ao médico de família por um dos membros da família mais próximos (a prima). Foi, dessa forma, a família que detectou os primeiros sinais e elaborou até as primeiras hipóteses diagnósticas.
De acordo com o princípio da autonomia do utente, este recorreu mais tarde ao seu médico de família, após ter sido confrontado com a preocupação dos seus familiares. Mais uma vez, a família foi determinante neste caso clínico, desta vez ao nível da influência sobre as crenças e atitudes do doente em relação à sua própria saúde. Nestas patologias, além do doente muitas vezes não notar as alterações iniciais, insidiosas, pode inclusive adaptar-se a esses movimentos involuntários, incorporando-os em movimentos voluntários. Dessa forma, podem minimizar a interferência com a sua autonomia funcional e/ou imagem social.
Este caso clínico permite também alertar para a patologia neurológica que cursa com movimentos involuntários anormais e para a forma mais correcta de a abordar. É uma avaliação e diagnóstico fundamentalmente clínico. Os exames auxiliares de diagnóstico são poucos e limitados na sua utilidade, pelo menos numa fase inicial. Eles servem sobretudo para a aplicação na investigação etiológica de apenas algumas das etiologias específicas de cada tipo de alteração do movimento e são do âmbito de actuação da especialidade de neurologia.
Neste caso, o parkinsonismo foi considerado um dos diagnósticos diferenciais, além da coreia, pela história pouco clara contada acerca do pai do doente. O parkinsonismo é uma alteração do movimento diferente: o tremor. O tremor difere da coreia por ser um movimento involuntário com um ritmo e amplitude regulares, não sendo abrupto e arrítmico como a coreia. No parkinsonismo, o tremor é tipicamente “de repouso”, sendo eliminado pelos movimentos voluntários do segmento afectado, geralmente extremidades distais, queixo e língua.17 Na doença de Parkinson existe ainda rigidez muscular, bradicinésia e marcha parkinsónica, tudo características não encontradas tipicamente na coreia, como neste caso.
Além do diagnóstico diferencial de parkinsonismo, que se punha pela história contada pelo próprio doente e pelos seus familiares, devem-se considerar as causas secundárias de coreia, as mais frequentes causas desta alteração do movimento. Na consulta de neurologia foram excluídas essas patologias, mediante exames complementares à avaliação clínica do paciente. Lá excluíram-se causas reumáticas, incluindo lúpus eritematoso sistémico (doente assintomático, com hemograma, marcadores inflamatórios e auto-anticorpos sem alterações de relevo), tirotoxicose (doente assintomático, com TSH e T4 livre normais), infecções (serologias de VIH 1 e 2 negativas), hiperglicemia e policetemia vera (hemograma e glicose normais). O restante estudo apresentava-se também sem alterações, apoiando a exclusão de outras causas, como síndromas paraneoplásicas. As causas iatrogénicas, sendo das mais frequentes, estavam à partida excluídas pela inexistência de medicação habitual (levodopa, neurolépticos, fenitoína, etc.) ou de drogas de abuso (cocaína). A idade de início e o início insidioso eram características a favor de uma causa primária, nomeadamente a doença de Huntington. A deterioração cognitiva, que também é característica da doença avançada, ainda não se verificava de forma objectiva (Mini-Mental State Test com resultado não significativamente diminuído), apesar da impressão de esquecimento por parte dos familiares. Além disso, a imagiologia (diminuição dos núcleos caudados) era compatível, apesar de não diagnóstica por si só. O estudo genético (resultado ainda não disponível à data de redacção do caso clínico) é o estudo laboratorial que confirma formalmente a doença. De notar a possibilidade remota (cerca de 1%) de, estando este doente diagnosticado clinicamente com a doença de Huntington, apresentar um teste genético negativo para a alteração genética da doença. Estes casos raros são formados no seu conjunto por fenocópias da doença, causadas por outras patologias hereditárias, as quais se pesquisam nesses casos (e.g., coreia hereditária benigna, Huntington disease-like syndromes, neuroacantose, doença de Wilson, etc.).6,8 Tratando-se de uma doença hereditária autossómica dominante, foi desta forma feito o diagnóstico provável, post mortem, do pai do doente. Além disso, a hereditariabilidade da doença levanta uma questão (frequente nesta doença) que é a manifestação clínica ser posterior à produção de descendência. Isto pode pôr em causa o aconselhamento genético atempado, como neste caso, em que o doente já possuía dois filhos de cinco anos de idade, tendo teoricamente 50% de probabilidade de ter transmitido o gene responsável pela doença. O estudo genético dos familiares directos do indivíduo afectado, neste caso aplicável aos descendentes do doente, permite saber o seu estado: se se tratam de indivíduos não portadores do gene e, por isso, “saudáveis” ou se herdaram o gene do pai, sendo portadores do gene e, por isso, futuros doentes, agora em fase pré-clínica da doença de Huntington. Apesar das questões éticas que este tipo de estudos pode levantar, e que não são do âmbito da discussão deste trabalho, o conhecimento do estado genético dos descendentes do doente, seja ele investigado na presente infância ou no futuro, por exemplo em fase pré-concepcional, permitirá, no caso de serem portadores da mutação e mesmo que a doença ainda não se tenha manifestado, decidir pelo recurso a métodos de reprodução medicamente assistida que evitem a transmissão do gene à descendência ou a métodos de diagnóstico pré-natal, com vista ao conhecimento do estadio de portador/não-portador do feto, existindo possibilidade da decisão pela interrupção da gravidez.
Neste caso é também possível a reflexão científica acerca da origem da doença de Huntington nesta família. Sendo o pai do doente o primeiro elemento conhecido provavelmente afectado pela doença, apresentam-se três cenários: o da origem do gene em pelo menos um dos avós paternos do doente, tendo manifestado a doença; o da origem do gene em pelo menos um dos avós paternos do doente, não tendo manifestado a doença, seja porque não tenham vivido anos suficientes para que se manifestasse, seja por serem portadores de um gene alterado que não se manifestou (entre 36 a 39 repetições da sequência genética CAG, alteração essa com penetrância incompleta, com a possibilidade de se ter expandido para 40 ou mais repetições - penetrância completa - nas duas gerações seguintes); ou ainda a mutação ter surgido espontaneamente pela primeira vez no pai do doente, não existindo nas gerações anteriores.
No que se refere ao tratamento e acompanhamento do doente, ele está actualmente medicado pelo neurologista com uma dose baixa, optimizada, de um neuroléptico (haloperidol), conseguindo na fase inicial da doença um bom controlo dos sintomas motores (coreia) da doença. Tendo em conta o curso natural da doença, será necessário um acompanhamento do doente, quer pelo neurologista quer pelo seu médico de família, no sentido da optimização de um plano que permita o melhor controlo possível dos sintomas não só motores, que progressivamente se agravarão, como da deterioração cognitiva e sintomas psiquiátricos que virão previsivelmente a aparecer e a progredir. Esse acompanhamento e controlo de sintomas será essencial para o próprio doente, no sentido de melhorar a sua qualidade de vida, progressivamente diminuída pela doença e para os seus conviventes e/ou cuidadores, no sentido de controlar o melhor possível o impacto negativo que o acto de cuidar lhes pode provocar. A institucionalização é uma das estratégias aplicáveis em fases avançadas da doença, quando o controlo farmacológico e fisioterapêutico já não é suficiente em ambulatório e/ou exista burnout dos cuidadores.
Concluindo, o presente trabalho aborda uma das doenças que cursam com movimentos involuntários anormais, alertando para a correcta abordagem médica. Como grande grupo, são doenças neurológicas comuns. A história clínica (onde podem ser relevantes os sinais notados por familiares/conviventes) e o exame físico (com alterações típicas no exame neurológico) são o principal método de diagnóstico dessas doenças.8,14,16 A posição que o médico de família ocupa no acesso aos cuidados de saúde coloca-o na posição de ser o primeiro a detectar os sintomas e sinais deste tipo de doenças que, no seu global, são das mais frequentes da área da neurologia. A detecção e a avaliação clínica inicial destas doenças estão ao seu alcance, referenciando os casos detectados aos cuidados de saúde secundários. Dessa forma, também intervém na gestão do processo de controlo destas doenças, articulando os dois níveis de cuidados. Neste caso clínico, o seguimento do utente ao longo da sua vida e o conhecimento e consideração do seu meio familiar e social constituíram ferramentas diagnósticas dificilmente acessíveis aos cuidados de saúde secundários.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Klein C. Movement disorders: classifications. J Inherit Metab Dis. 2005;28(3):425-39. [ Links ]
2. Walker HK, Hall WD, Hurst JW. Clinical methods: the history, physical, and laboratory examination. 3rd ed. Boston: Butterworths; 1990. [ Links ] ISBN 9780409900774
3. Bhidayasiri R, Truong D. Chorea and related disorders. Postgrad Med J. 2004;80(947):527-34. [ Links ]
4. Richards RI. Dynamic mutations: a decade of unstable expanded repeats in human genetic disease. Hum Mol Genet. 2001;10(20):2187-94. [ Links ]
5. Conneally PM. Huntington disease: genetics and epidemiology. Am J Hum Genet. 1984;36(3):506-26. [ Links ]
6. Suchowersky O, Bouchard M. Overview of chorea (Monografia na Internet). Waltham, MA: UpToDate; 2014 (updated 2015 May 12; cited 2014 Aug 13). Available from: http://www.uptodate.com/contents/overview-of-chorea
7. Shannon KM. Treatment of chorea. Continuum (Minneap Minn). 2007;13(1):72-93. [ Links ]
8. Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis. 2010;5:40. [ Links ]
9. Walker FO. Huntington's disease. Lancet. 2007;369(9557):218-28. [ Links ]
10. Costa MC, Magalhães P, Ferreirinha F, Guimarães L, Januário C, Gaspar I, et al. Molecular diagnosis of Huntington disease in Portugal: implications for genetic counselling and clinical practice. Eur J Hum Genet. 2003;11(11):872-8. [ Links ]
11. Jones HR, Burns TM, Aminoff MJ, Pomeroy SI. Colecção Netter de ilustrações médicas (Internet). 2ª ed. Rio de Janeiro: Elsevier; 2014. ISBN 9788535274691. Available from: https://issuu.com/elsevier_saude/docs/e-sample_jones_ii [ Links ]
12. Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60(5):1202-10. [ Links ]
13. Suchowersky O, Muthipeedika J. A case of late-onset chorea. Nat Clin Pract Neurol. 2005;1(2):113-6. [ Links ]
14. Piccolo I, Defanti CA, Soliveri P, Volontè MA, Cislaghi G, Girotti F. Cause and course in a series of patients with sporadic chorea. J Neurol. 2003;250(4):429-35. [ Links ]
15. Reed TE, Chandler JH, Hughes EM, Davidson RT. Huntington’s chorea in Michigan: I. Demography and genetics. Am J Hum Gen. 1958;10(2):201-25. [ Links ]
16. McCusker EA, Gunn DG, Epping EA, Loy CT, Radford K, Griffith J, et al. Unawareness of motor phenoconversion in Huntington disease. Neurology. 2013;81(13):1141-7. [ Links ]
17. Chou KL. Diagnosis of Parkinson disease (Monografia na Internet). Waltham, MA: UpToDate; 2014 (updated 2016 Feb 29; cited 2014 Aug 13). Available from: http://www.uptodate.com/contents/diagnosis-of-parkinson-disease
Endereço para correspondência | Dirección para correspondencia | Correspondence
Hugo Taveira Cunha
Moinhos Velhos - Bravães, 4980-124 Ponte da Barca
E-mail: hugotaveiradacunha@outlook.com
Conflito de interesses
Os autores declaram não ter conflitos de interesses.
Recebido em 27-05-2015
Aceite para publicação em 08-02-2016

