








Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAngiologia e Cirurgia Vascular
versión impresa ISSN 1646-706X
Angiol Cir Vasc vol.14 no.1 Lisboa mar. 2018
CASO CLÍNICO
Patients with new SDHD gene mutation with carotid body paragangliomas
Nova mutação do gene SDHD em pacientes com paragangliomas do corpo carotídeo
Roger Rodrigues1, Maria Almeida2, João Carreiro3, Carolina Mendes1, Juliana Varino1, André Marinho1, Bárbara Pereira1, Mário Moreira1, Mafalda Botelho1, Óscar Gonçalves1, Albuquerque Matos1
1Serviço de Angiologia e Cirurgia Vascular, Centro Hospitalar e Universitário de Coimbra
2 Centro de Neurociências e Biologia Celular, Universidade de Coimbra
3 Serviço de Cirurgia Maxilofacial, Centro Hospitalar e Universitário de Coimbra
Autor para correspondência
ABSTRACT
Paragangliomas (PGLs) are neuroendocrine neoplasms that can occur throughout the body wherever there is paraganglia. Representing 0.03% of all tumours, PGLs are extremely rare. Although predominantly benign and amenable to cure by surgical ressection, up to 6% can be malignant. To date, three genes have been identified that are associated with Familial PGLs. All three encode subunits (D, B and C) of the enzyme succinate dehydrogenase complex (SDH), which is part of the Kreb's cycle and the electron transport chain. We report a novel causative frameshift mutation in the subunit D of SDH (SDHD) in a family with Carotid Body Paragangliomas. This finding contributes for extending the known mutational spectrum of SDHD, and to help the genetic counseling of this family. Noteworthy, is now possible to offer to other relatives, still sub-clinical, a predictive test that would eventually aid an early surveillance/intervention for a better prognosis.
Keywords: Carotid body paraganglioma; Familial paraganglioma; Mutation; SDHD; Succinate dehydrogenase; Surgery.
RESUMO
Introdução: Os paragangliomas (PGLs) são neoplasias neuroendócrinas que podem ocorrer em todo o corpo onde exista paraganglia. Representando 0,03% de todos os tumores, os PGLs são extremamente raros.
Embora predominantemente benignos e passíveis de cura por ressecção cirúrgica, estima-se que até 6% possam ser malignos. PGLs familiares têm um modo de transmissão autossômico dominante, e uma variabilidade fenotípica significativa. Até ao momento, foram identificados três genes associados aos PGLs familiares. Todas as subunidades de codificação (D, B e C) do complexo enzima succinato desidrogenase (SDH), fazem parte do ciclo de Krebs e da cadeia transportadora de electrões.
Objectivos: Pretende-se descrever uma nova frameshift mutation no gene SDHD identificado numa família com Paragangliomas do Corpo Carotídeo.
Resultados: A análise de mutação do probando revelou a presença de uma nova frameshift mutation, c.549delG (p.L139Ffs), no exão 4 do gene SDHD. Ambos os descendentes herdaram essa mutação patogênica, embora num deles ainda esteja subclínico.
Conclusão: Descrevemos uma nova mutação causal de frameshift na subunidade D do SDH (SDHD) numa família com Paragangliomas do Corpo Carotídeo. Esta descoberta contribui para o conhecimento do espectro mutacional do SDHD, e para ajudar o aconselhamento genético desta família. Destacamos o facto de agora ser possível oferecer a outros familiares, ainda subclínicos, um teste preditivo que permite auxiliar uma vigilância ou intervenção precoce, e consequentemente melhorar o prognóstico.
Palavras-chave: Paraganglioma; Tumor do corpo carotídeo; Paraganglioma familiar; Mutação; SDHD; Succinato desidrogenase; Cirurgia.
Paragangliomas (PGLs) are neuroendocrine neoplasms that can occur throughout the body wherever there is paraganglia, non-neuronal cells derived from the neural crest so named for being in close proximity to sympathetic ganglia. There are fundamentally two types of paraganglia: chromaffin or sympathetic made of chromaffin cells, which have primary endocrine functions and non-chromaffin or parasympathetic made of glomus cells, which have primary chemoreceptor functions.(1)
Representing 0.03% of all tumours, PGLs are extremely rare. Although predominantly benign and amenable to cure by surgical resection, up to 6% can be malignant.(2) About 50% are found in the head and neck region, most commonly as highly vascularised carotid body tumors, which represent approximately 65% of head and neck PGLs.(3)(4) PGLs can also be found as aortic glomus, jugular bulb and vagal and tympanic nerves. Depending on their location, patients with carotid body PGLs may notice a slow growing and painless lateral neck mass, pulse-like sensations or voice changes.
Three different types of Carotid Body PGLs have been described in the literature: Sporadic, Familial and Hyperplastic. The Sporadic form is by far the most common type, representing approximately 85% of these tumors. The prevalence of the Familial type has varied between reported studies from as low as 5% to as high as 50%.(5) This wide-ranging variability stems from the existence of hidden familial cases. In the latter, diagnosis is usually made earlier (30-35 years) and there's a higher prevalence of bilateral multifocal PGLs.(5) The Hyperplastic form is very common in patients with chronic hipoxia, such as those in New Mexico, Peru and Colorado who live at a high altitude (>1524 meters/5000 feet above sea level) and in patients with chronic obstructive pulmonary disease (COPD) or cyanotic heart disease.(6)
Familial PGLs have an autosomal dominant mode of inheritance, therefore all generations of a family can be affected. Nevertheless, due to their exceedingly high phenotypic variability, very different clinical manifestations can occur in patients with the same genotype, even among family members. Genetic heterogeneity, incomplete penetrance and genetic imprinting lie at the root of this phenotypic variability. To date, three genes have been identified that are associated with Familial PGLs. All three encode subunits (D, B and C) of the enzyme succinate dehydrogenase complex (SDH), which is part of the Kreb's cycle and the electron transport chain. Defective succinate dehydrogenase has been postulated to cause an increase in the intra
CASE PRESENTATION
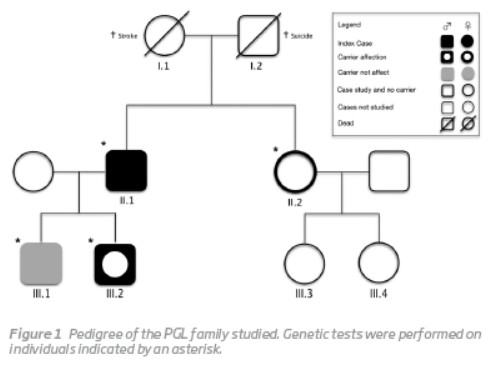
The family pedigree is presented in Figura 1.
INTRODUCTION
The index case (II.1) was a 58-year-old man with a known history of bilateral PGL of the carotid body. The diagnosis had been established 35 years earlier when he first presented to the Vascular Surgery outpatient clinic for evaluation of a left latero-cervical mass. Angiography suggested a carotid body PGL that was surgically removed through subadventitial dissection. A similar mass was detected in the contralateral side 9 years later and it was also removed through subadventitial dissection. In both cases, the paraganglioma diagnosis was confirmed histologically. The current mutation analysis was conducted in the context of a systematic review of paraganglioma cases operated in the Vascular Surgery Department of Centro Hospitalar e Universitário de Coimbra (CHUC). DNA was isolated from peripheral blood using the DNA isolation kit and all the coding region of the SDHD gene (1-4 exons) was PCR amplified. The presence of the pathogenic mutation was always confirmed on a separate amplification with subsequent direct sequencing. The analysis revealed that the patient harboured a novel frameshift mutation, c.549delG (p.L139Ffs), in exon 4 of the SDHD gene. The proband's father (I.2) committed suicide when he was 76 years old while his mother (I.1) died of stroke. Both their DNAs were not available for testing. His sister (II.2) underwent genetic testing but the mutation was not found.
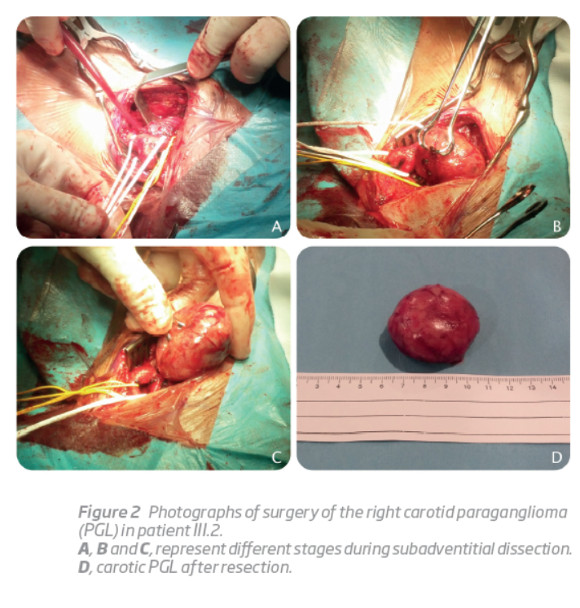
His younger son (III.2), a 23-year-old man with otherwise irrelevant past medical history, was referred to the Vascular Surgery outpatient clinic because of a painless right latero-cervical mass with progressive growth during the previous 6 months. Physical examination revealed a regular and mobile mass with approximately 6 cm of diameter, in the projection of the right carotid triangle, anterior to the sternocleidomastoid muscle and immediately above the hyoid bone. A bruit was also present. A computerized tomography angiography was ordered, suggesting a carotid body PGL compressing right carotid vessels. Selective angiography was performed 2 days before surgical removal of the tumor, and the feeding artery was successfully embolized with coils. A subadventitial tumor excision was then performed (Figura 2). The histological examination showed highly vascular tissue and clusters of Zellballen cells. The subsequent mutation analysis of SDHD exon 4 showed that he also carried the family mutation.
The proband's eldest son (III.1, age 28 years) has also been found to carry the mutation, although at the present he remains sub-clinical. His sister, a 60-year-old woman, and her two daughters don't display any signs or symptoms suggestive of PGL.No other case was detected in the family.
DISCUSSION
In the present study, the mutation analysis for SDHD gene has been performed in a family with PGLs of the carotid body. An heterozygous mutation, c.549delG; p. L139Ffs in the SDHD gene has been identified in the index case and in his two offsprings (one still unaffected). This frameshift mutation was novel, not previously described (http://hgmd.cf.ac.uk) and was located in exon 4 of the SDHD gene (Figura 3).
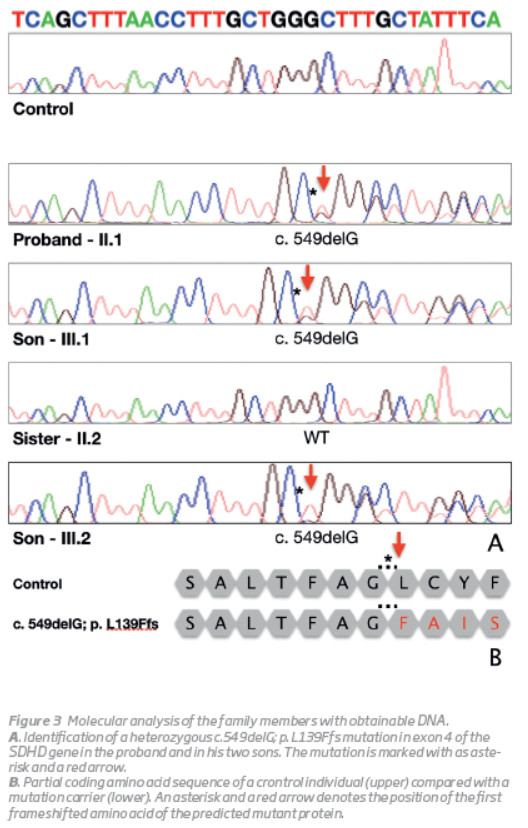
The genetic testing for this one base pair deletion was extended to the sister of the index patient and in accordance with her phenotype, no mutation has been found. As illustrated in this family, PGLs should always be considered in patients presenting with swelling at the lateral neck, cranial nerve palsies, voice changes or auditory defects. A detailed family history and thorough clinical examination are of paramount importance. Ultrasound of soft tissue and vessels of the neck is mandatory. MR angiography should also be performed.(9)
The successful management of PGLs hinges on early diagnosis. The familial type has been associated with age at diagnosis <40 years, bilateral multifocal tumors, preceding pheochromocytoma and PGL family history. That said, due to the disease's striking phenotypic variability, individuals with Familial PGLs can erroneously be clinically ascertained as having sporadic appearing PGLs.(10)(11) Furthermore, it has been reported than up to 30% of apparently sporadic PGLs are caused by germline mutations.(12) Familial PGLs are thus underdiagnosed. Diagnosis is often established when a patient seeks clinical advice because of a palpable mass. At that stage, the tumor is often large enough to pose significant intra-operatory challenges. Since the only cure for PGLs is complete surgical excision(10), early diagnosis is critical for the operative outcome. The incidence of additional vascular procedures and nerve lesions has in fact been reported to be significantly lower in mutation carriers.(9) The timely detection of both unilateral and contralateral PGLs in these patients enabled them to be operated upon earlier, which translated into less surgical complications, consequence of the more favourable operative outcome associated with smaller tumors.
Mutation carriers should be screened clinically and radiologically. Ultrasound and additional MR angiography scans of the head and neck regions, thorax, abdomen and pelvis should be performed seeing that PGLs can occur throughout the entire vegetative system.(10) Genetic testing should be extended to children. Accordingly to the European-American Paraganglioma Study Group, screening should start at 10 years of age, with follow-up screens every 2 years.(13) The tumor's sluggish growth, with doubling times varying from 4 and 14 years(9), obviates the need for annual examinations. Non-carrier family members don't need to be screened.
Population-specific variations in the prevalence, penetrance, and phenotypic expression of SDH subunit gene mutations have been postulated. The well documented association of SDHD mutations with head and neck PGLs may in fact vary across different populations. Further genetic studies are necessary.
We report a family with a novel heterozygous frameshift mutation in the SDHD gene, expanding the clinical and genetic heterogeneity of hereditary PGLs. This study highlights the importance of mutation analysis for individual follow-up strategy, in particular for mutation carriers who should undergo regular clinical examination. This procedure will certainly provide an early detection of PGLs in a context of genetic counseling and thereby improve the operative outcome of these patients.
REFERENCES
1. Anne Marie McNicol (2010). "Chapter 12: Adrenal medulla and paraganglia". Endocrine Pathology: Differential Diagnosis and Molecular Advance (Springer ed.). p. 281. [ Links ]
2. Kruger AJ, Walker PJ, Foster WJ, et al. Important observations made managing carotid body tumours during a 25 year experience. J Vasc Surg 2010;52:1518e23. [ Links ]
3. Rao AB, Koeller KK, Adair CF. From the archives of the AFIP. Paragangliomas of the head and neck: radiologic-pathologic correlation. Radiographics 1999;19:1605e32. [ Links ]
4. Georgiadis GS, Lazarides MK, Tsalkidis A, et al. Carotid body tumor in a 13-year-old child: Case report and review of the literature. J Vasc Surg. 2008 Apr. 47(4):874-880. [Medline]. [ Links ]
5. Fakhry N, Niccoli-Sire P, Barlier-Seti A, et al. Cervical paragangliomas: is SDH genetic analysis systematically required? Eur Arch Otorhinolaryngol 2008;265:557–63. [ Links ]
6. Sajid MS, Hamilton G, Baker DM. A multicenter review of carotid body tumour management. Eur J Vasc Endovasc Surg. Aug/2007. 34:127-30. [Medline]. [Full Text]. [ Links ]
7. Sevilla Garcia MA, Llorente Pendas JL, Rodrigo Tapia JP, et al. [Head and neck paragangliomas: revision of 89 cases in 73 patients]. Acta Otorrinolaringol Esp. 2007 Mar. 58(3):94-100. [Medline]. [ Links ]
8. Baysal BE. Genomic imprinting and environment in hereditary paraganglioma. Am J Med Genet C Semin Med Genet 2004;129C:85–90. [ Links ]
9. Fruhmann J, Geigl JB, Konstantiniuk P, et al. Paraganglioma of the Carotid Body: Treatment Strategy and SDH-gene Mutations. Eur J Vasc Endovasc Surg. May/2013. 45:431-36. [ Links ]
10. Boedeker CC. Paragangliome und Paragangliomsyndrome. Laryngo-Rhino-Otol 2011;90:S56e82. [ Links ]
11. De Toma G, Nicolanti V, Plocco M, et al. Baroreflex failure syndrome after bilateral excision of carotid body tumors: an underestimated problem. J Vasc Surg 2000;31:806e10. [ Links ]
12. Boedeker CC, Neumann HP, Offergeld C, et al. Clinical features of paraganglioma syndromes. Skull Base 2009;19:17e25. [ Links ]
13. Myssiorek D, Ferlito A, Silver CE, et al. Screening for familial paragangliomas. Oral Oncol 2008;44:532–7. [ Links ]
Correio eletrónico: roger.cc@hotmail.com (R. Rodrigues).
Recebido a 10 de maio de 2017
Aceite a 07 de maio de 2018