









Services on Demand
Journal
Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Portuguesa de Ortopedia e Traumatologia
Print version ISSN 1646-2122On-line version ISSN 1646-2939
Rev. Port. Ortop. Traum. vol.26 no.1 Lisboa Mar. 2018
CASO CLÍNICO
Doença de Camurati-Engelman - A propósito de um caso clínico
Sandra SantosI; Vítor Hugo PinheiroI; André PintoI; Marcos CarvalhoI; António Pais LopesI; Fernando FonsecaI
I. Serviço de Ortopedia, Centro Hospitalar e Universitário de Coimbra. Coimbra.
RESUMO
Objetivo: O presente caso clínico tem como objetivo apresentar o tratamento, resultado clínico e imagiológico de uma lesão traumática (fratura supracondiliana do fémur) num doente com doença de Camurati-Engelmann, doença displásica óssea que produz deformidade hiperostótica da diáfise dos ossos longos. A doença de Camurati-Engelmann é uma doença genética autossómica dominante rara, de prevalência desconhecida No entanto, dos vários casos e famílias descritas na literatura, 12 pertencem a uma família portuguesa, o que torna o caso, bem como o conhecimento da doença e sua patologia, relevantes. De igual modo, não estão descritos na literatura casos de tratamento cirúrgico de patologia traumática nestes doentes, o que torna este caso particularmente interessante.
Descrição: Trata-se de um doente de 46 anos, que sofreu fratura supracondiliana do fémur após acidente de motociclo. A opção terapêutica escolhida para o tratamento da fratura foi cirúrgica, com osteossíntese da fratura com placa anatómica bloqueada. O resultado clínico aos 2 anos de pós-operatório foi excelente, tendo o doente regressado às suas atividade de vida diárias sem restrições, e com boa mobilidade; o estudo imagiológico mostrou igualmente consolidação da fratura.
Comentários: A relevância deste caso assenta na descrição do tratamento e resultado funcional após fratura supracondiliana do fémur num doente com doença de Camurati-Engelmann, situação rara que os autores não encontraram abordado na literatura atual.
Palavras chave: Doença de Camurati-Engelmann, Fratura, Tratamento cirúrgico.
ABSTRACT
Objective: This clinical case report intends to present the treatment, clinical and radiological outcomes of a traumatic injury (supracondylar femur fracture) in a patient with Camurati-Engelmann disease, bone dysplasia that produces a hyperostotic deformity of the long bone diaphysis. Camurati-Engelmann’s disease is a rare autossomic dominant genetic disease, of unknown prevalence. However, among the various sporadic cases and families described in the literature, 12 patients belong to a Portuguese family, which makes this case report, as well as knowledge of the disease and its pathology, relevant. Likewise, there are no reports on the surgical management of traumatic injuries in this patient population in the current literature, which makes this case particularly interesting.
Description: A 46-year old male presented in the Emergency Room with a supracondylar femur fracture, after motorcycle accident. The chosen treatment was surgical, with open reduction and osteossynthesis with an anatomical locked plate. Clinical result at 2 years post-operatively was favorable, with no restrictions on activities of daily living and good range of motion; x-ray studies showed fracture consolidation.
Comments: The relevance of the present report is based on the description of the treatment and functional outcome after supracondylar femur fracture in a patient with Camurati-Engelmann disease, a rare presentation which the authors have not found in current literature.
Key words: Camurati-Engelmann disease, Fracture, Surgical treatment.
INTRODUÇÃO
A doença de Camurati-Engelmann (DCE), também designada displasia diafisária progressiva, é uma doença de transmissão genética autossómica dominante, com expressividade variável, pertencendo a um grupo mais amplo de patologias, as hiperostoses craniotubulares. A primeira descrição clínica desta doença data de 1920, tendo sido Camurati, em 1922, o primeiro a sugerir a sua natureza hereditária1,2.
A mutação genética responsável pela doença localiza-se no gene TGFB1, que codifica o fator de crescimento transformador ß1 (FCTß1), uma molécula que, em condições fisiológicas, estimula a formação óssea e suprime a reabsorção. Todas as mutações descritas na literatura, até à data, mostram um aumento de atividade do FCTß12,3.
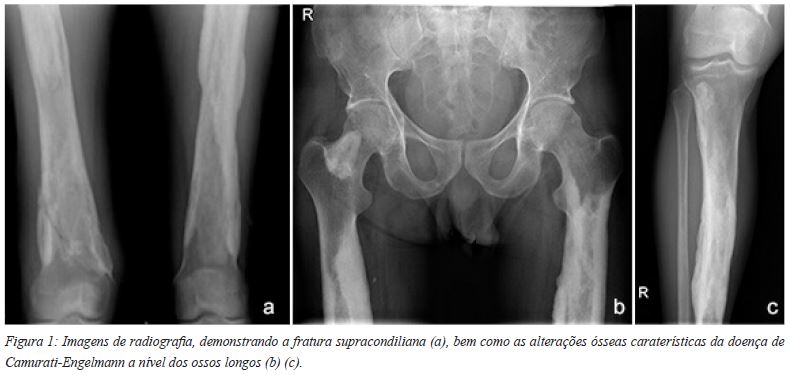
A principal característica clínica desta doença é o espessamento da cortical nas diáfises dos ossos longos, habitualmente bilateral e simétrica, observando-se inicialmente no fémur e tíbia e, mais tarde e à medida que a doença progride, também na fíbula, úmero, ulna e rádio. As regiões metafisárias podem também ser atingidas, mas as epífises são sempre poupadas2,4,5.
A doença manifesta-se, por norma, durante a infância e adolescência, com uma apresentação clínica caracterizada por dor nos membros inferiores, fraqueza muscular, alterações da marcha e cansaço fácil. Manifestações sistémicas podem também estar presentes, tais como anemia, leucopenia e hepatoesplenomegália. Analiticamente, é possível observar níveis aumentados dos marcadores do metabolismo ósseo2,5.
O tratamento passa, sobretudo, pelo tratamento médico, com recurso a fármacos como corticosteróides, bifosfonatos, calcitonina e anti-inflamatórios não esteróides, tendo como principal objetivo o alívio da dor. Existem relatos de tratamento cirúrgico através de rimagem do canal medular, baseando-se na premissa de que o aumento da pressão intra-medular provocado pelo aumento da espessura cortical e redução do espaço intra-medular seria responsável pelas queixas álgicas, o que nunca foi demonstrado, carecendo de fundamentação e evidência científica. A terapia genética poderá ser, no futuro, uma alternativa para o tratamento desta doença; no entanto, este tipo de terapêutica encontra-se ainda nos seus estadios iniciais de desenvolvimento2.
A literatura disponível sobre esta doença é limitada, e a grande maioria dos trabalhos publicados debruça-se sobre os aspectos genéticos e moleculares da doença, bem como as suas caraterísticas clínicas e imagiológicas2,3.
CASO CLÍNICO
Trata-se de doente do sexo masculino, 46 anos, que se apresentou no Serviço de Urgência por gonalgia à direita com incapacidade funcional, após traumatismo (acidente de motociclo). O exame objetivo mostrou derrame intra-articular a nível do joelho direito, com dor à mobilização passiva. O estudo radiológico mostrou uma fratura supra-condiliana do fémur direito (Figura 1(a)) (33-A1, segundo a classificação AO), onde são evidentes as alterações ósseas induzidas pela doença (Figura 1(b) e (c)).
Foi realizada uma artrocentese, com drenagem de cerca de 40cc de conteúdo hemático com glóbulos de gordura.
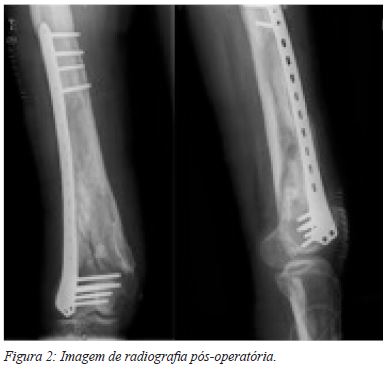
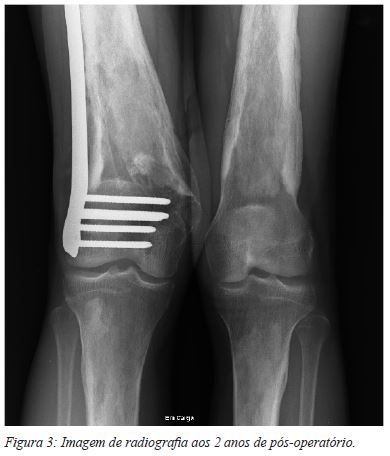
Optou-se pelo tratamento cirúrgico, por técnica minimamente invasiva e osteossíntese com placa anatómica bloqueada (Figura 2). No período pós-operatório, iniciou imediatamente mobilização passiva assistida e ativa, sendo permitida a deambulação precoce com apoio de canadianas (apenas com contato ao solo durante as primeiras 4 semanas, e depois com carga progressiva conforme tolerado). O último follow-up do doente, aos 2 anos de pós-operatório, revelou boa consolidação da fratura (Figura 3). Clinicamente, já tinha retomado as suas atividades de vida diária, com as limitações prévias inerentes à patologia de base, sem dor e sem necessidade de apoios de marcha, apresentando uma flexão de 120º com extensão de 0º e um ligeiro valgo, sem repercussão funcional.
DISCUSSÃO
A DCE é uma doença rara, cuja prevalência é desconhecida (estimam-se cerca de 200 casos a nível mundial). Estão descritas na literatura algumas dezenas de famílias atingidas pela doença2, bem como alguns casos esporádicos6; não foi encontrado pelos autores, até ao momento, nenhum caso descrevendo o tratamento de lesões traumáticas. Curiosamente, uma das famílias com maior números de casos identificados, localiza-se em Portugal (12 elementos da família com doença conhecida)2,7,8, o que torna este caso relevante, pela maior probabilidade de encontrar casos de doença na nossa prática clínica.
Apesar de bem descritas na literatura as alterações ósseas caraterísticas desta patologia, não existem relatos sobre a resposta destes doentes a lesões traumáticas, como fraturas. Tal fato torna difícil a comparação com diferentes tipos de tratamento e resultados funcionais.
No presente caso, verificou-se um bom resultado funcional e uma adequada consolidação da fratura. É sabido que o FCTß1 tem um efeito positivo na consolidação óssea9. O conhecimento da patofisiologia da doença, o aumento de atividade do FCTß1 e consequente estímulo da atividade osteoblástica e inibição da atividade osteoclástica poderia fazer presumir o aparecimento de um calo ósseo exuberante, ou mesmo conduzir a alterações da consolidação, como pseudartrose hipertrófica. Tal não se verificou, o que sugere que a atuação do FCTß1 poderá ser condicionada por outros fatores locais, como citocinas e outros mediadores inflamatórios, relacionados com o processo de fratura e consolidação.
Há também a considerar a menor morbilidade da técnica minimamente invasiva como um fator favorecedor de um melhor resultado funcional, mas como referido previamente, a ausência de outros casos reportados torna difícil essa comparação.
Os autores consideram, assim, que em casos de fratura em doentes com doença de Camurati-Engelmann, o tratamento cirúrgico deverá ser utilizado, dando primazia ao uso de técnicas minimamente invasivas.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Sparkes RS, Graham CB. Camurati-Engelmann Disease Genetics and Clinical Manifestations with a Review of the Literature. J Med Genet. 1972 Mar; 9 (1): 73-85
2. Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006 Jan; 43 (1): 1-11
3. Cohen MM Jr. The new bone biology: Pathologic, molecular, and clinical correlates. Am J Med Genet A. 2006 Dec 1; 140 (23): 2646-2706
4. Uezato S, Dias G, Inada J, Valente M, Fernandes E. Imaging aspects of Camurati-Engelmann disease. Rev Assoc Med Bras. 2016; 62 (9): 825-827 [ Links ]
5. Bartuseviciene A, Samuilis A, Skucas J. Camurati-Engelmann disease: imaging, clinical features and differential diagnosis. Skeletal Radiol. 2009; 38: 1037-1043 [ Links ]
6. Grey AC, Wallace R, Crone M. Engelmann's Disease: a 45-year follow-up. J Bone Joint Surg Br. 1996; 78-B: 488-491 [ Links ]
7. Almeida J, Beça G, Laíns J. Acta Reumatol Port. 2012; 37: 122-126 [ Links ]
8. Garcia J, Monteiro P, Saavedra MJ, Silva J, Malcata A. Doença de Camurati-Engelmann. Acta Reumatol Port. 2007; 32: 395-396 [ Links ]
9. Poniatowski LA, Wojdasiewicz P, Gasik R, Szukiewicz D. Transforming Growth Factor Beta Family: Insight into the Role of Growth Factors in Regulation of Fracture Healing Biology and Potential Clinical Applications. Mediators Inflamm. 2015; 2015 [ Links ]
Conflito de interesse:
Nada a declarar
Sandra Santos
Serviço de Ortopedia dos Hospitais da Universidade de Coimbra
Praceta Prof. Mota Pinto
3000-075 COIMBRA
Telefone: 239 400 400
sandrafs_86@hotmail.com
Data de Submissão: 2017-10-06
Data de Revisão: 2018-01-08
Data de Aceitação: 2018-01-08

{kind=link}