








Servicios Personalizados
Revista
Articulo
 Portugués (pdf)
Portugués (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Portuguesa de Ortopedia e Traumatologia
versión impresa ISSN 1646-2122
Rev. Port. Ortop. Traum. vol.20 no.4 Lisboa dic. 2012
CASO CLÍNICO
Osteomielite xantogranulomatosa do úmero
Rui NunesI; Jorge CostaI; Manuela MartinsI
I. Serviço de Ortopedia. Hospital de S. José. Centro Hospitalar de Lisboa Central. Lisboa. Portugal.
RESUMO
Os autores apresentam um caso de lesão osteolítica da metáfise distal do úmero em que o exame anatomopatológico permitiu estabelecer o diagnóstico de osteomielite xantogranulomatosa.
Esta entidade nosológica pouco frequente, que pode mimetizar patologia óssea tumoral, é uma forma de osteomielite crónica caracterizada histologicamente pela acumulação de histiócitos vacuolizados e de células inflamatórias.
Os autores descrevem neste artigo a histopatologia deste tipo de osteomielite e discutem o seu diagnóstico diferencial.
Palavras chave: Osteomielite xantogranulomatosa, células xantomatosas, histiócitos.
ABSTRACT
The authors present a case of an osteolytic lesion of the distal metaphysis of the humerus in wich the histopathological examination established the diagnosis of xanthogranulomatous osteomyelitis.
This uncommon nosological entity, wich can mimic bone tumor, is a form of chronic osteomyelitis histologically characterized by the accumulation of foamy histiocytes and inflammatory cells.
The authors describe the histopathology of this type of osteomyelitis and discuss its differential diagnosis.
Key words: Xanthogranulomatous osteomyelitis, foam cells, histiocytes.
INTRODUÇÃO
A inflamação xantogranulomatosa é uma forma de reação inflamatória crónica na qual se verifica a acumulação tecidual de numerosos histiócitos vacuolizados (células espumosas ou xantomatosas), de leucócitos e de plasmócitos[1]. Este tipo de processo inflamatório pode manifestar-se em diversos orgãos, apresentando uma particular incidência no rim e na vesícula biliar. A sua ocorrência a nível ósseo tem sido contudo muito raramente descrita, encontrando-se referenciados apenas cinco casos na literatura.
A osteomielite xantogranulomatosa foi descrita pela primeira vez em 1984 por A. Cozzutto[2]; este autor menciona dois casos, com localizações na tíbia e na primeira costela. Os outros três casos publicados apresentavam as seguintes localizações: cúbito, úmero e peróneo (no mesmo doente) e tíbia[3,4,5].
CASO CLÍNICO
Uma mulher de 56 anos, de raça negra, recorreu em dezembro de 2011 ao serviço de urgência do nosso hospital por queixa de dor localizada a nível do cotovelo direito, com duas semanas de evolução; negava a ocorrência de traumatismo como fator desencadeante.
À observação apresentava discreto edema da região posterior do cotovelo e mantinha esta articulação em moderada flexão, sendo que a sua mobilização ativa ou passiva exacerbava a dor. O restante exame objetivo era normal.
A doente encontrava-se a realizar terapêutica antiretroviral, apresentando seropositividade para o vírus da imunodeficiência humana conhecida desde 2007.
O exame radiológico convencional do cotovelo evidenciou a existência de lesão osteolítica localizada na metáfise distal do úmero, de contorno irregular, com as dimensões máximas de 26 e 15 milímetros nas incidências antero-posterior e de perfil, respetivamente. Observava-se disrupção da cortical óssea anterior e não existia reação perióstea (Figura 1).
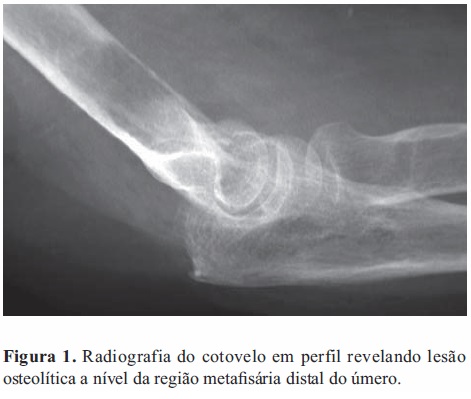
Foi colocada a hipótese diagnóstica de tumor ósseo (primário ou metastático).
O estudo morfológico da lesão lítica umeral realizou-se por tomografia axial computorizada, que não revelou compromisso das partes moles envolventes (Figura 2). As cavidades torácica, abdominal e pélvica foram avaliadas pelo mesmo método, não tendo sido encontradas lesões sugestivas de patologia neoplásica.
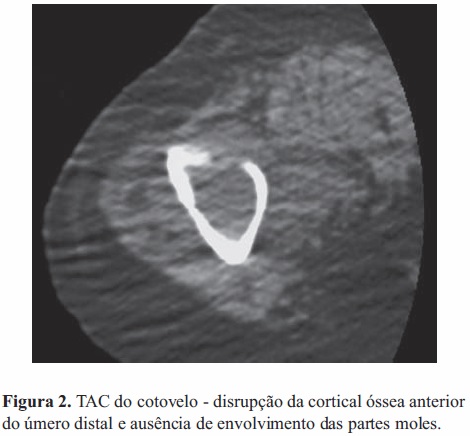
A cintigrafia óssea de corpo inteiro com tecnésio evidenciou hipercaptação do radiofármaco na região distal do úmero direito; e heterogeneidade de fixação em ambos os ombros e no joelho direito, sendo este último padrão compatível com a presença de alterações degenerativas osteoarticulares a estes níveis.
Os exames laboratoriais revelaram leucocitose (18.400 leucócitos/mL) com neutrofilia (fórmula leucocitária com 92,9 % de neutrófilos). Existia elevação da proteína C reativa (98,6 mg/L). Os doseamentos séricos de fosfatase alcalina e de cálcio encontravam-se dentro dos limites da normalidade.
A doente foi submetida a intervenção cirúrgica na qual se obteve acesso à lesão osteolítica do úmero distal por via posterior transtricipital, com isolamento do nervo cubital.
Constatou-se a presença, a nível da região metafisária distal do úmero, de uma massa de consistência mole, de coloração branco-amarelada, que foi excisada na sua totalidade por curetagem. A cavidade óssea resultante foi preenchida com pasta biocerâmica de hidroxiapatite. Procedeu-se a estabilização óssea com duas placas retas de pequenos fragmentos, colocadas uma em cada coluna do úmero distal.
O exame anatomopatológico do material colhido intraoperatoriamente revelou tratar-se de tecido inflamatório constituído predominantemente por histiócitos, muitos dos quais de aparência xantomatosa; identificaram-se também numerosos neutrófilos e alguns linfócitos e plasmócitos; não foram observadas células neoplásicas (Figura 3).
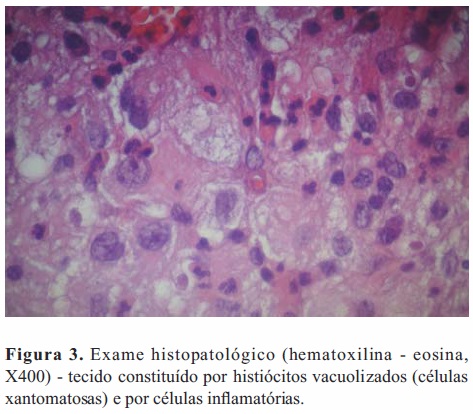
Com o objetivo de caracterizar melhor as células xantomatosas procedeu-se ao seu estudo por métodos imunocitoquímicos. Foi assim pesquisada a presença do marcador CD68, cujo resultado positivo permitiu identificar estas células como histiócitos (Figura 4); e do marcador CD1a, cujo resultado negativo permitiu excluir que se tratassem de células de Langerhans.
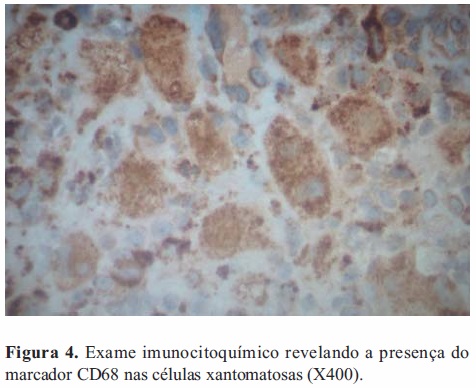
Foi ainda realizada a pesquisa de citoqueratinas nas células da amostra, utilizando-se para o efeito os anticorpos anticitoqueratinas AE1 e AE3; o resultado foi negativo, confirmando que não se estava na presença de metástase de carcinoma.
O conjunto destas observações permitiu efetuar o diagnóstico de osteomielite xantogranulomatosa.
O pós-operatório decorreu sem complicações, não tendo contudo a doente cumprido o programa de fisioterapia prescrito.
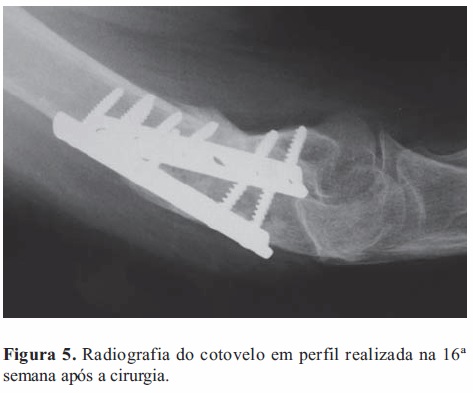
Na avaliação realizada na 16ª semana após a cirurgia a doente apresentava-se sem dor e sem edema do cotovelo; observava-se limitação de 18º da extensão desta articulação e não havia limitação da flexão.
O exame radiológico do cotovelo mostrou normalização da densidade óssea a nível da região metafisária distal do úmero e restauração da continuidade da cortical anterior; o material de osteossíntese permanecia corretamente implantado (Figura 5).
Nesta mesma data foram solicitados hemograma e doseamento da proteína C reativa, tendo a doente recusado a realização destes exames.
DISCUSSÃO
A inflamação óssea xantogranulomatosa é uma forma rara de osteomielite crónica cujas manifestações clínicas e radiográficas podem mimetizar patologia óssea tumoral. O diagnóstico definitivo apenas pode ser estabelecido pelo exame histopatológico, caracterizando-se a inflamação xantogranulomatosa pela acumulação tecidual de histiócitos vacuolizados em conjunto com outras células inflamatórias (leucócitos, plasmócitos e células gigantes multinucleadas).
Os histiócitos são células do sistema monocítico-macrofágico, ou seja, são macrófagos com origem nos monócitos do sangue circulante. Estas células, que apresentam uma ampla distribuição em múltiplos tecidos do organismo, participam na reação inflamatória e estão implicadas na resposta imunitária pelo seu papel como células apresentadoras de antigénios. Os histiócitos são dotados da capacidade de fagocitose, sendo os materiais fagocitados acumulados em vacúolos do citoplasma. Os histiócitos vacuolizados, também designados por células espumosas ou xantomatosas, são macrófagos modificados pela fagocitose de partículas lipídicas.
As células xantomatosas são células volumosas e geralmente mononucleadas; o seu citoplasma , abundante, apresenta-se distendido por vacúolos.
Na patogenia da inflamação xantogranulomatosa das partes moles têm sido implicados diversos fatores como a infeção, a necrose e a hemorragia teciduais. No entanto, nenhum destes fatores foi até hoje demonstrado como estando na origem da osteomielite xantogranulomatosa.
O diagnóstico diferencial deste tipo peculiar de osteomielite deve ter em consideração todas as outras entidades nosológicas que podem cursar com acumulação de histiócitos em um ou mais pontos do esqueleto. Para efeitos de sistematização classificámos estas entidades em três grupos: histiocitoses, tumores ósseos histiocitários e doenças de armazenamento lisossómico (Quadro I).
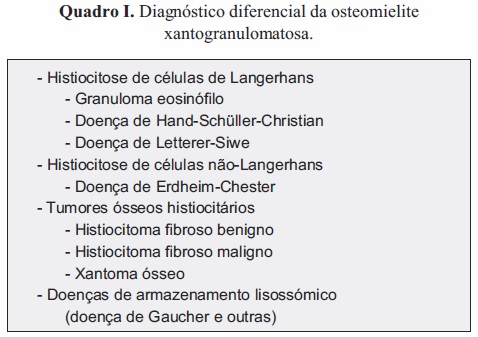
As histiocitoses são um conjunto de doenças relativamente pouco frequentes, de patogenia ainda não completamente esclarecida e cujo espetro de apresentação clínica é amplo.
A histiocitose de células de Langerhans (denominada anteriormente histiocitose X) engloba três entidades nas quais ocorre proliferação de células de Langerhans em vários tecidos: o granuloma eosinófilo, a doença de Hand-Schüller-Christian e a doença de Letterer-Siwe.
A mais frequente destas patologias é o granuloma eosinófilo. Esta doença apresenta maior incidência nas primeiras três décadas de vida. Manifesta-se como lesão lítica mono ou, mais raramente, poliostótica; embora estas lesões sejam mais frequentes nos ossos planos do crâneo e da bacia, nas vértebras e nas costelas, podem também surgir nos ossos longos do esqueleto apendicular. Histologicamente a doença caracteriza-se por proliferação de células de Langerhans, de macrófagos espumosos e de leucócitos, havendo geralmente predominância de eosinófilos.
As células de Langerhans são um tipo particular de histiócitos. São células volumosas, com um ou mais núcleos de contorno irregular; têm reduzidos sinais de atividade fagocitária, embora possam apresentar-se como células xantomatosas por acumulação de lípidos em vacúolos do citoplasma.
O diagnóstico definitivo de histiocitose de células de Langerhans pode ser estabelecido pelo método imunocitoquímico, sendo que as células de Langerhans são imunoreativas para o marcador CD1a; ou por exame ultraestrutural por microscopia eletrónica, caracterizando-se estas células pela presença de estruturas citoplasmáticas denominadas grânulos de Birbeck.
No caso que descrevemos, o exame histológico não demonstrou a presença de eosinófilos e a pesquisa do marcador CD1a nas células histiocitárias do tecido colhido intraoperatoriamente revelou-se negativa, pelo que foi excluído o diagnóstico de granuloma eosinófilo.
As doenças de Hand-Schüller-Christian e de Letterer-Siwe apresentam aspetos histológicos similares aos do granuloma eosinófilo. São contudo formas disseminadas com envolvimento multiorgânico, caracterizando-se ainda por serem exclusivas da infância.
A doença de Erdheim-Chester é uma histiocitose de células não-Langerhans que se manifesta a partir da 5ª década de vida. A doença decorre com envolvimento esquelético multifocal e, em cerca de metade dos casos, extraesquelético. O exame radiográfico revela tipicamente esclerose óssea bilateral e simétrica envolvendo os ossos longos dos membros.
Os histiocitomas fibrosos (benigno e maligno) são tumores pouco frequentes que podem ocorrer em diversos pontos do organismo, inclusivamente no esqueleto. Ambos os tumores são constituídos por proliferação de histiócitos, que podem apresentar aparência xantomatosa, associados a numerosas células fibroblásticas.
O xantoma ósseo é uma lesão pseudotumoral rara. Esta doença, que em alguns casos se associa a hiperlipidemia, manifesta-se como lesão osteolítica constituída no exame histológico quase exclusivamente por macrófagos distendidos por acumulação lipídica; o infiltrado inflamatório não é significativo.
Finalmente, devem ser consideradas no diagnóstico diferencial as doenças de armazenamento lisossómico (como a doença de Gaucher), uma vez que estas entidades nosológicas, que têm múltiplas formas de apresentação, podem cursar com proliferação de células de aparência xantomatosa na medula óssea. O mecanismo patogénico consiste na acumulação citoplasmática de biomoléculas devido a deficiência de enzimas lisossómicas.
O reduzido número de casos publicados e a nossa experiência com um único caso não permitem estabelecer conclusões definitivas sobre o tratamento e o prognóstico da osteomielite xantogranulomatosa. A curetagem de todo o tecido anómalo complementada pelo preenchimento da cavidade óssea resultante parece no entanto permitir obter a cura da doença. A ocorrência de fratura patológica poderá determinar a necessidade de recorrer a métodos de osteossíntese.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Cozzutto C, Carbone A. The xanthogranulomatous process. Xanthogranulomatous inflammation. Pathol Res Pract. 1988; 183: 395-402 [ Links ]
2. Cozzutto C. Xanthogranulomatous osteomyelitis. Arch Pathol Lab Med. 1984; 108: 973-976 [ Links ]
3. Vankalakunti M, Saikia U, Mathew M, Kang M. Xanthogranulomatous osteomyelitis of ulna mimicking neoplasm. World J Surg Oncol. 2007; 5: 46 [ Links ]
4. Borjian A, Rezaei F, Eshaghi M, Shemshaki H. Xanthogranulomatous osteomyelitis. J Orthopaed Traumatol. 2011; 13 (4): 217-220 [ Links ]
5. Kamat G, Gramapurohit V, Myageri A, Shettar C. Xanthogranulomatous osteomyelitis presenting as swelling in right tibia. Case Reports in Pathol. 2011; 2011 [ Links ]
Conflito de interesse:
Nada a declarar.
Rui Nunes
Rua das Caravelas, lote 4.45.01C – 5ºB
1990-608 Lisboa
Portugal
rnunes65@gmail.com
Data de Submissão: 2012-05-30
Data de Revisão: 2012-10-27
Data de Aceitação: 2012-11-05