










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Ortopedia e Traumatologia
versão impressa ISSN 1646-2122
Rev. Port. Ortop. Traum. vol.20 no.1 Lisboa mar. 2012
CASO CLÍNICO
Síndrome do tunel cárpico bilateral em criança com mucopolissacaridose tipo VI
Rui PimentaI, II; Nuno AlegreteI, II; Sara LimaI, II; Elisa TelesI, II; Esmeralda RodriguesI, II; Gilberto CostaI, II
I. Serviço de Pediatria. Unidade de Doenças Metabólicas. Centro Hospitalar de São João. Porto. Portugal.
II. Serviço de Ortopedia Infantil. Centro Hospitalar de São João. Porto. Portugal.
RESUMO
O síndrome do túnel cárpico é raro em crianças. O grupo das mucopolissacaridoses constitui, na população pediátrica, a causa mais frequente deste quadro clínico. Com o advento das novas terapias para o tratamento específi co de alguns tipos de mucopolissacaridoses, o seu prognóstico alterou-se significativamente, com diminuição de morbilidade e aumento da sobrevida do doente, assumindo as manifestações músculo-esqueléticas ainda maior relevo na abordagem global do doente. Desta forma, o diagnóstico e tratamento atempado do síndrome do túnel cárpico nestes doentes torna-se fundamental para a obtenção de bons resultados funcionais e melhoria da qualidade de vida. Os autores apresentam um caso clínico de uma doente de 12 anos com mucopolissacaridose tipo VI e síndrome do túnel cárpico bilateral, submetida a tratamento cirúrgico no mesmo tempo operatório.
Palavras chave: síndrome do túnel cárpico, idade pediátrica, mucopolissacaridose.
ABSTRACT
Carpal tunnel syndrome in children is uncommon. Mucopolysaccharidosis is the most common cause of carpal syndrome in this age group. With the new treatment available for mucopolysaccharidosis, the progression of this disease has greatly decreased, and all its musculoskeletal manifestations now assume more importance in the global treatment of these patients. Early diagnosis and treatment of carpal tunnel syndrome in these children are important to achieve good functional outcome. The authors present a case report of bilateral carpal tunnel syndrome in a 12 year-old patient, operated at once.
Key words: Carpal tunnel syndrome, paediatric age, mucopolysaccharidosis.
INTRODUÇÃO
O síndrome do túnel cárpico (STC) é a neuropatia compressiva mais comum em adultos, contudo raramente está presente na infância e na adolescência. Neste grupo etário, as mucopolissacaridoses (MPS) constituem a causa mais frequente desta patologia.
CASO CLÍNICO
Doente do sexo feminino, com 12 anos, seguida em consulta de Pediatria desde o segundo ano de vida por atraso de crescimento, tendo sido diagnosticada MPS tipo VI (Doença de Maroteaux-Lamy). Aos 7 anos avaliada em centro terciário, sendo registado fenótipo com envolvimento multissistémico grave e iniciou terapêutica enzimática de substituição com galsulfase aos 8 anos de idade. Por quadro compressivo medular efetuou, em 2009, descompressão medular cervical, com melhoria do estado geral, nomeadamente de cefaleias e do padrão de marcha.
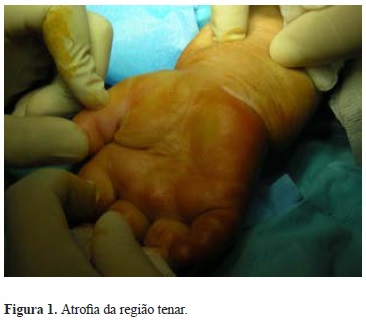
Desde o inicio de 2010, referia parestesias de predomínio matinal a nível do território do nervo mediano em ambas as mãos, sem artralgias. Ao exame clínico notou-se progressiva atrofia da região tenar bilateralmente (Figura 1), diminuição generalizada das amplitudes articulares nos punhos e nas mãos e sinal de Tinel positivo bilateralmente. Efetuou eletromiografia com resultados compatíveis com síndrome do túnel cárpico bilateral.
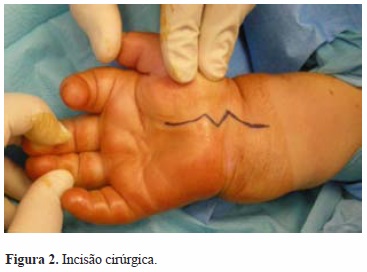
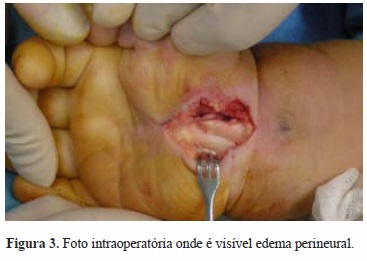
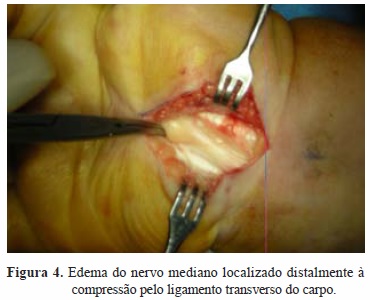
Foi submetida a descompressão cirúrgica do nervo mediano bilateralmente, no mesmo tempo cirúrgico (Figuras 2 e 3). Intraoperatoriamente constatou-se a presença de edema perineural do nervo mediano bilateral, distalmente à área de compressão pelo ligamento transverso do carpo, que, conjuntamente com as bainhas dos tendões flexores, se apresentava espessado (Figuras 4 e 5).
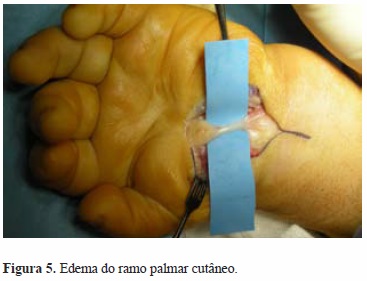
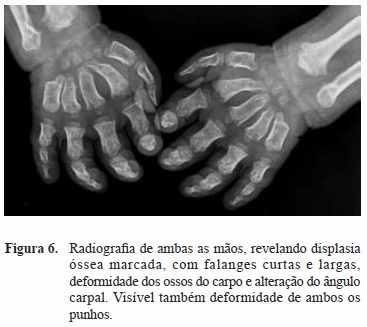
Com este procedimento houve remissão completa do quadro clínico, com melhora imediata dos sintomas. Um ano depois a doente recuperou a sensibilidade e recuperou progressivamente alguma capacidade de oponência dos polegares, mantendo as limitações associadas à displasia óssea característica da doença (Figura 6).
DISCUSSÃO
As mucopolissacaridoses (MPS) são doenças metabólicas hereditárias raras, integradas no grupo das doenças de sobrecarga lisossomal, devidas a deficiência de enzimas que atuam na degradação dos glicosaminoglicanos (GAG) – anteriormente denominados mucopolissacáridos -, açúcares complexos com importantes funções celulares, nomeadamente a nível de processos de adesão e sustentação. Na ausência da hidrolase lisossomal específica, ocorre deposição progressiva dos GAG não catabolizados a nível de diferentes tecidos, nomeadamente com repercussões a nível do tecido conjuntivo na pele, válvulas cardíacas, vias aéreas e esqueleto. O armazenamento de GAG causa disfunção celular e, a nível macroscópico, alteração dos tecidos e órgãos. Conforme a enzima defi ciente e o substrato acumulado, são identificados 7 tipos diferentes de MPS. As suas manifestações clínicas variam, sendo normalmente multissistémicas, com apresentações fenotípicas diversas desde formas leves a muito graves da doença [1].
A MPS tipo VI é devida à defi ciência da enzima N-acetilgalactosamina-4-sulfatase, resultando na alteração do catabolismo do sulfato de dermatano. Estima-se que afete cerca de 1100 pessoas em todo o mundo, sendo referida uma incidência aparentemente mais elevada no norte de Portugal (Quadro I)[2,3]. O fenótipo clínico descrito pela primeira vez por Maroteaux-Lamy em 1963, apresenta manifestações clínicas semelhantes à MPS tipo I (Doença de Hurler), com uma exceção importante: o desenvolvimento cognitivo em geral não é afetado nos doentes acometidos por MPS tipo VI.

À medida que aumenta a quantidade de GAG acumulado, a apresentação clínica da doença torna-se progressivamente mais severa. Os primeiros sinais são geralmente alterações pediátricas frequentes, tais como otites, infeções respiratórias recorrentes, atraso de crescimento e a existência de uma hérnia umbilical ou inguinal. Progressivamente vai ocorrendo o envolvimento multissistémico característico da doença: baixa estatura, dismorfia facial progressiva, opacidade da córnea, otite média recorrente e hipoacusia, rinossinusite, pneumonia, valvulopatia cardíaca e cardiomiopatia, hepatoesplenomegalia, rigidez articular e alterações esqueléticas características que no seu conjunto constituem a Disostosis Multiplex. Em praticamente todos os doentes o resultado final é semelhante, com progressão da doença, tornando os indivíduos cada vez mais debilitados e consequente diminuição da esperança de vida.
Com o advento das novas abordagens terapêuticas, nomeadamente transplante de medula óssea e administração de enzima recombinante específica (disponível atualmente para as MPS I, II e VI), tem-se registado um melhor controlo da progressão da doença a nível cardáco e respiratório, com consequente aumento da qualidade de vida dos doentes. Quanto às manifestações musculo-esqueléticas, embora a terapia enzimática de substituição iniciada no primeiro anos de vida em doentes com MPS tipo VI pareça ter um efeito muito positivo, na maior parte dos casos não se verificou uma melhoria significativa, tendo estas ganho maior importância. Encontra-se mesmo descrito que o tratamento com transplante de células hematopoiéticas pode agravar o quadro ortopédico devido à acumulação de GAG específicos na cartilagem, tendões e cápsula articular [4].
As manifestações ortopédicas mais frequentes na MPS são STC, dedo em mola, cifose toracolombar, joelho valgo, displasia da anca, diminuição generalizada da ossificação encondral com implicação no crescimento ósseo e rigidez articular generalizada[5].
As doenças metabólicas de sobrecarga, como as MPS são a causa mais frequente STC em crianças e adolescentes, tendo muitas vezes associada a presença de dedos em mola. A evolução do síndrome do túnel cárpico nestes doentes ocorre devido ao espessamento fibroso do ligamento transverso do carpo e das restantes bainhas tendinosas, com envolvimento dos tecidos adjacentes despoletado pelo depósito constante e progressivo de GAG, seguindo-se o edema perineural com a consequente desmielinização nervosa [6].
Clinicamente apresenta-se com dor a nível do punho ou mão, provocando por vezes despertar noturno, recusa do paciente em usar a mão acometida e hipersensibilidade ao toque, diminuição da destreza manual fina e atrofia muscular, principalmente na região tenar. Por vezes a criança rói as unhas tentando desesperadamente aliviar o desconforto que sente. Sintomas típicos do STC como parestesias e disestesias raramente são descritos pela natural dificuldade de comunicação e expressão da criança. O sinal de Tinel ou de Phalen estão normalmente ausentes.
Tratando-se de uma entidade de diagnóstico fundamentalmente clínico, neste contexto nem sempre é fácil o seu diagnóstico assumindo a eletromiografia (EMG) um papel fundamental na sua deteção.
Para além de uma história clínica e familiar cuidadosa, todos os doentes com MPS, mesmo assintomáticos, devem ser submetidos a uma avaliação precoce por uma equipa multidisciplinar e ponderar a realização de EMG para avaliar a condução nervosa dos nervos mediano e cubital a nível do punho. Mesmo doentes assintomáticos poderão apresentar alterações na condução nervosa detetadas apenas por este exame [5]. Nestes casos, deve manter-se vigilância atenta, não havendo indicação para cirurgia profi lática. Caso os doentes refiram sintomas compatíveis com STC e apresentem alterações electromiográficas, devem ser de imediato submetidos a descompressão cirúrgica do nervo mediano, no sentido de diminuir a probabilidade de instalação de lesões irreversíveis.
Para promover uma libertação eficaz do nervo mediano, para além da secção do ligamento transverso, é por vezes necessário efetuar tenossinovectomia no sentido de reduzir o volume de tecido ao nível do canal cárpico. Apesar do espessamento do epineuro, não está demonstrado que a epineurectomia efetuada conjuntamente, melhore o resultado funcional destes doentes[7,8]. Caso algum dedo em mola esteja presente, este deve ser submetido a libertação da polia e, se necessário, ressecção parcial do tendão flexor superficial dos dedos, no sentido de maximizar o potencial de recuperação da funcionalidade da mão no pós-operatório.
Um programa de fisioterapia intensivo após a libertação cirúrgica do nervo mediano contribui para uma melhoria do resultado funcional nestes doentes contudo, os melhores resultados obtêm-se em indivíduos sem deficiência cognitiva associada, devido à sua melhor compreensão e colaboração no pós-operatório, tal como no caso apresentado.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Neufeld EF, Muenzer J. The mucopolysaccharidoses. In Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. p. 3421-3452.
2. Meikle PJ, Hopwood JJ, Clague AE. Prevalence of lysosomal storage disorders. JAMA. 1999; 281: 249-254 [ Links ]
3. Pinto R, Caseiro C, Lemos M. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. 2004; 12: 87-92 [ Links ]
4. Khanna G, Van-Heest AE, Agel J. Analysis of factors affecting development of carpal tunnel syndrome in patients with Hurler syndrome after hematopoietic cell transplantation. Bone Marrow Transplantation. 2007; 39: 331-334 [ Links ]
5. Wraith JE, Alani SM. Carpal tunnel syndrome in the mucopolysaccharidoses and related disorders. Arch Dis Child. 1990; 65: 962-963 [ Links ]
6. Al-Qatan MM, Thomson HG, Clark HM. Carpal tunnel syndrome in children and adolescents with no history of trauma. J Hand Surg [Br]. 1996; 21B: 108-111 [ Links ]
7. Blair WF, Goetz DD, Ross MA. Carpal tunnel release with or without epineurectomy: a comparative prospective trial. J Hand Sur [Am]. 1996; 21: 655-661 [ Links ]
8. Borisch N, Haussmann P. Neurophysiological recovery after open carpal tunnel decompression with epineurectomy. J Hand Surg [Br]. 2003; 28: 450-454 [ Links ]
Conflito de interesse:
Nada a declarar.
Rui Pimenta
Serviço de Ortopedia
Hospital São João ? FMUP
Al. Prof. Hernâni Monteiro
4200 Porto
Portugal
ruipimentaribeiro@gmail.com
Data de Aceitação: 2011-08-12

