










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Pneumologia
versão impressa ISSN 0873-2159
Rev Port Pneumol v.16 n.3 Lisboa jun. 2010
Tumor maligno da bainha dos nervos periféricos do pulmão: A propósito de um caso clínico
N Serrano Marçal1, E Teixeira2, R Sotto-Mayor3, A Manique4, P Campos5, J Cruz6, M Mendes de Almeida7, A Bugalho de Almeida8
1 Interno do internato complementar de Pneumologia*
2 Assistente hospitalar graduada de Pneumologia*
3 Chefe de serviço de Pneumologia*
4 Assistente hospitalar graduada de Pneumologia*
5 Assistente hospitalar graduada de Radiologia†
6 Assistente hospitalar graduado de Cirurgia Cardiotorácica‡
7 Assistente hospitalar graduada de Anatomia Patológica§
8 Chefe de serviço de Pneumologia*
* Serviço de Pneumologia 1
† Serviço de Imagiologia Geral 1
‡ Serviço de Cirurgia Cardio-Torácica
§ Serviço de Anatomia Patológica
Hospital de Santa Maria, CHLN, EPE, Lisboa
Resumo
Os tumores malignos da bainha dos nervos periféricos correspondem a um grupo raro de sarcomas de tecidos moles que tendem a ocorrer em doentes com neurofibromatose tipo 1 ou vários anos após tratamentos de radioterapia. A sua localização torácica é uma entidade muito rara. A sintomatologia habitual deve-se ao efeito de massa exercido sobre os nervos, podendo persistir durante meses ou anos antes do diagnóstico. A escassez de doentes com este tipo de tumor faz com que a abordagem terapêutica ainda permaneça em debate, sendo a cirurgia o procedimento de eleição. O prognóstico destes doentes é mau dada a grande recorrência local e metastização. O caso clínico descrito serve de tema à discussão.
Palavras-chave: Tumor maligno, bainha nervo periférico.
Malignant peripheral nerve sheath tumors: A case report
Abstract
Malignant peripheral nerve sheath tumors comprehend a rare group of soft tissue sarcomas that tend to occur in patients with neurofibromatosis type 1 or several years after radiotherapy treatments. Its thoracic localization is a very unusual entity. The typical symptoms are due to nerve roots compression which can persist for several months or years before diagnosis. Due to very few patients with this type of tumor its therapeutic approach is still a matter of permanent debate, being surgery the main treatment. This tumor has a bad prognosis because of high local recurrence and metastasis. The clinical case we describe serves as a glimpse for discussion.
Key-words: Malignant tumour, peripheral nerve sheath.
Introdução
Os sarcomas correspondem a um grupo heterogéneo de neoplasias, dos quais os tumores malignos da bainha dos nervos periféricos (MPNST) fazem parte. Outrora também conhecidos como schwannomas malignos, neurofibrossarcomas, sarcomas neurogéneos ou neurilemomas malignos, a actual designação de MPNST parece ser a que maior consenso reúne na comunidade médica1. Tal deve-se ao facto de a célula de origem destes tumores não estar ainda completamente esclarecida, admitindo-se contudo que a maioria advém das células de Schwann1,2. Estes tumores tendem a ocorrer sobretudo nas extremidades e só muito raramente ocorrem na cavidade torácica3.
Dada a particularidade destas neoplasias, sobretudo a nível torácico, com consequente limitação na obtenção de estudos com dados significativos referentes à sua abordagem, apresentamos o presente caso clínico para motivar a discussão sobre estes tumores e a actuação clínica mais adequada ao seu tratamento.
Caso clínico
JRM, sexo masculino, raça negra, 31 anos, camionista, natural do Brasil e residente em Portugal há oito anos. Encontrava-se assintomático até quatro meses antes do internamento no serviço, altura em que iniciou queixas de perda ponderal (cerca de 10 kg) e cansaço. Cerca de duas semanas antes, referiu o aparecimento de tosse seca de agravamento progressivo, odinofagia, disfonia e toracalgia posterior esquerda, contínua, sem relação com os movimentos respiratórios e que aliviava em decúbito lateral esquerdo. Negava outros sintomas. Automedicou-se com diclofenac, paracetamol, ambroxol e amoxicilina sem melhoria, motivo pelo que recorreu ao serviço de urgência do nosso hospital.
Como antecedentes pessoais há apenas a referir que se tratava de um não fumador, sem hábitos toxicofílicos e não utilizava qualquer terapêutica farmacológica regular.
O exame objectivo revelava um doente com bom estado geral, vigil, acianótico, eupneico, hemodinamicamente estável, apirético e com uma saturação de oxigénio em ar ambiente de 95%. Não apresentava adenomegalias palpáveis e as mucosas eram coradas e hidratadas. A auscultação cardíaca não apresentava alterações. Da avaliação do tórax ressaltava a abolição das vibrações vocais e macicez na percussão do hemitórax esquerdo e na auscultação pulmonar o murmúrio vesicular estava abolido também à esquerda e não eram audíveis ruídos adventícios. A avaliação do abdómen e membros era normal. Analiticamente mostrava uma discreta anemia (Hb=11,8 g/dL) normocítica mas hipocrómica (HGM=26,8 pg) e aumento dos parâmetros inflamatórios (VS=67 mm; PCR=5,7 mg/dL), ainda que sem leucocitose. A gasometria arterial em ar ambiente era normal e a radiografia do tórax (Fig. 1) demonstrava uma volumosa massa com preenchimento de praticamente todo o campo pulmonar esquerdo, condicionando desvio do mediastino para a direita e uma obliteração do seio costo frénico esquerdo sugestiva de derrame pleural. Ainda no serviço de urgência realizou ecografia torácica que demonstrou moderado derrame pleural à esquerda, com aspectos sugestivos de loculação. O doente ficou internado para estudo.
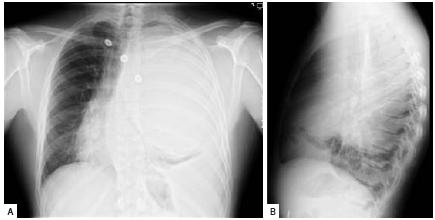
Fig. 1 – Telerradiografi a de tórax PA (A) e perfi l (B) à data do internamento
No internamento foi submetido a toracocentese com saída de cerca 500 ml de líquido hemático velho. As características do líquido pleural eram compatíveis com exsudado com predomínio de neutrófilos e tinha um pH de 7,032, motivo pelo que ficou com drenagem pleural contínua. Os exames bacteriológicos, micológicos e micobacteriológicos do líquido pleural foram negativos e o exame anatomopatológico mostrou-se negativo para células neoplásicas.
Realizou tomografia computorizada (TC) do tórax (Fig. 2) que revelou uma volumosa massa ocupando a metade superior do hemitórax esquerdo com captação heterogénea de contraste, não se visualizando o brônquio lobar superior nem o ramo esquerdo da artéria pulmonar; colapso parcial do LIE; derrame pleural à esquerda; desvio do mediastino para a direita; adenopatias em localização pré-traqueal e paracardíaca anterior. Posteriormente realizou uma broncofibroscopia, onde se evidenciava na árvore brônquica esquerda um esporão do lobo superior alargado, segmento apicoposterior diminuído de calibre e segmento anterior com uma massa gelatinosa. Na biópsia brônquica documentou-se neoplasia fusocelular de padrão neural.
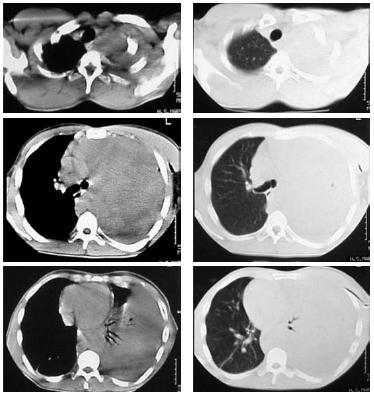
Fig. 2 – TC de tórax à data do internamento
Dos vários marcadores tumorais pedidos (á -fetoproteína, â2 -microglobulina, NSE, CA 15.3, CA 19.9, CEA, CYFRA -21 e SCC) apenas o NSE se encontrava aumentado (25,7 ug/L; VR<16,3 ug/L).
No decurso da marcha diagnóstica assistiu-se a um agravamento rápido do estado clínico, caracterizado por toracalgia refractária à terapêutica médica e aparecimento de dispneia em repouso, de agravamento progressivo. Radiologicamente apresentava um aumento do volume da massa, com maior desvio con-tralateral do mediastino (Fig. 3). Não havia evidência de doença extratorácica.
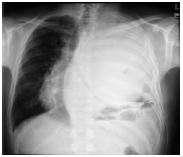
Fig. 3 – Telerradiografia de tórax PA duas semanas após o internamento
Por apresentar este agravamento clínico e imagiológico e se tratar de um doente jovem e sem diagnóstico definitivo, foi, em reunião multidisciplinar, proposto a cirurgia. Foi submetido a uma toracotomia esquerda de urgência, verificando -se que o lobo superior e segmentos do lobo inferior estavam destruídos pelo tumor (Fig. 4).
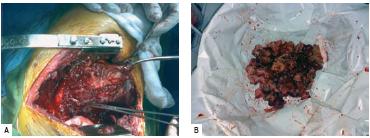
Fig. 4 – Destruição pulmonar pelo tumor (A). Massa tumoral ressecada (B)
Verificava-se hemorragia activa ao nível dos ramos apicais da artéria pulmonar, que se laquearam. Decidiu-se no procedimento por uma pneumectomia esquerda. Laqueação da artéria pulmonar esquerda e encerramento do brônquio principal. As veias pulmonares esquerdas estavam invadidas pelo tumor, tendo-se procedido à abertura do pericárdio, isolamento das veias pulmonares e clampagem da aurícula esquerda (AE). Ressecção em bloco das veias e parte da AE, que foi encerrada com “prolene 5:0” (Fig. 5).
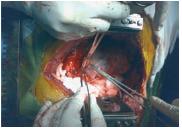
Fig. 5 – Encerramento das veias pulmonares superior e inferior a nível da AE
Finalmente, foram ressecados parcialmente o pericárdio e a pleura parietal, permitindo que do ponto de vista cirúrgico a ressecção tumoral fosse considerada completa (Fig. 6).
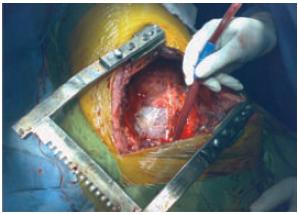
Fig. 6 – Aspecto final pós-pneumectomia radical com ressecção parcial do pericárdio e pleura parietal
O estudo morfológico da peça ressecada mostrou uma proliferação fusocelular, com padrão fasciculado (Fig. 7) e diferenciação neural, isto é, alternância abrupta entre áreas celulares e áreas mixóides (Fig. 8). Presença de necrose e 16 mitoses/10 CGA.

Fig. 7 – Neoplasia fusocelular, padrão fasciculado. H&E x 400
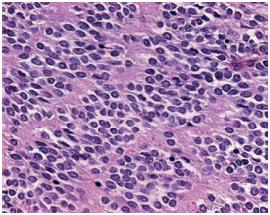
Fig. 8 – Diferenciação neural. H&E × 400
Observou-se imunorreactividade intensa e difusa para vimentina (Fig. 9) e positividade focal para S100 e EMA (Fig. 10). Negatividade para CD99, MNF116, CK7, calretinina, LCA, AML, CD34, miogenina e desmina.
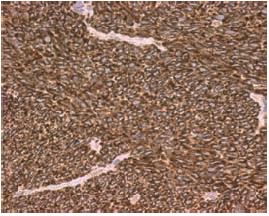
Fig. 9 – Positividade intensa e difusa para vimentina
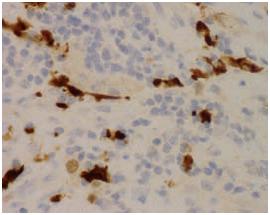
Fig. 10 – Imagem onde se evidencia o marcador neural S100 focalmente positivo
Os estudos de biologia molecular, realizados no Instituto Português de Oncologia de Francisco Gentil, em Lisboa, não revelaram alterações diagnósticas, nomeadamente a análise por FISH não revelou rearranjo dos genes EWS e SYT e a análise por CGH revelou a presença das seguintes alterações cromossómicas: ganho total do cromossoma 19 e ganhos parciais das regiões 13q21.2, 17q11.2-q12, 22q11.2, 22q13.1-q13.3. Considerou-se que se tratava de um sarcoma com padrão morfológico e fenotípico compatível com tumor maligno da bainha dos nervos periféricos. Do ponto de vista microscópico, havia igualmente infiltração da pleura parietal por tecido de neoplasia do mesmo tipo.
Perante o diagnóstico de sarcoma de subtipo MPNST em estádio anatómico III B e fisiológico com performance status 1 (ECOG), com tumor totalmente ressecado, o doente foi submetido a 4 ciclos de quimioterapia (QT) adjuvante com doxorrubicina e ifosfamida. O tratamento foi bem tolerado, tendo tido como efeitos secundários alopecia de grau 1 e episódios autolimitados de náuseas grau 1, que cederam à terapêutica antiemética. Como intercorrência há a referir que ao 8.º dia após o último ciclo o doente esteve internado três dias por neutropenia grau 4 associada a diarreia líquida. Foi medicado com G-CSF e terapêutica sintomática, com resolução do quadro clínico.
Posteriormente, e coincidindo com o reaparecimento de toracalgia à esquerda, realiza RM torácica, que demonstra loca pós-pneumectomia expandida por líquido não puro, e à periferia apical e lateral da cavidade observavam-se nódulos sólidos inferiores a 1 cm e que captavam o contraste injectado (Fig. 11).
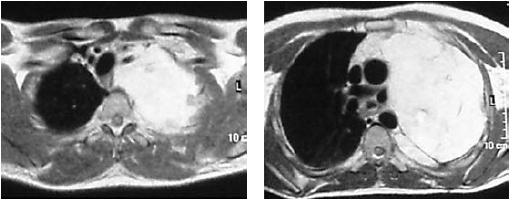
Fig. 11 – RM torácica onde se evidenciam nódulos à periferia
Iniciou tratamentos de radioterapia torácica externa com dose total de 66 Gy em 33 fracções, com melhoria da sintomatologia. A RM torácica de controlo documentou uma importante melhoria, quer do volume de líquido, quer em relação aos nódulos pleurais (Fig. 12).
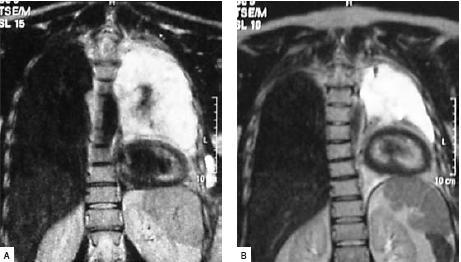
Fig. 12 – RM torácica realizada antes (A) e após (B) tratamentos de radioterapia
Actualmente o doente encontra-se em seguimento em consulta há 18 meses, estando assintomático.
Discussão
Segundo dados epidemiológicos dos EUA, a incidência dos tumores malignos da bainha dos nervos periféricos na população em geral é de 0,001%, o que corresponde entre 5 a 10% do total de sarcomas de tecidos moles diagnosticados anualmente2,4. Na maioria das séries verifica-se uma incidência igual nos dois sexos; contudo, em alguns estudos parece haver um ligeiro predomínio nas mulheres1,4. A idade de diagnóstico varia entre os 20 e os 50 anos, sendo que, nos doentes com neurofibromatose tipo 1, também conhecida como doença de von Recklinghausen, estes tumores geralmente são diagnosticados mais cedo1,4,5.
São conhecidos dois factores de risco para este tipo de tumor. O primeiro é a presença de neurofibromatose de tipo 1 (NF1), uma vez que têm um risco 4600 vezes maior de contrair estes tumores quando comparados com a população em geral4. Além disso, 50 a 60% dos MPNST ocorre em doentes com NF1. O segundo factor de risco conhecido são os tratamentos prévios de RT, sendo, nestes doentes, em regra diagnosticados após 15 anos do tratamento inicial2. A frequência da localização não varia com a existência ou não de NF14. Tal como nos restantes tumores neurogénicos, estes podem ocorrer em todo o tórax; no entanto, são encontrados habitualmente no mediastino posterior, no ângulo costovertebral6.
Os MPNST tendem a surgir como uma massa cujo aumento de dimensões é progressivo, a que se associam frequentemente sintomas, como dor e défices neurológicos sensoriais ou motores, secundários à compressão de um nervo1,4,7. Estes sintomas tendem a persistir durante meses ou anos5.
Em termos imagiológicos, a ressonância magnética é o exame de eleição, uma vez que tem a capacidade de distinguir os tumores de tecidos moles neurogénicos dos não neurogénicos1. No entanto, apesar de existirem algumas características que sugiram malignidade, este método tem limitações na sua distinção2,8. A tomografia computorizada mostra especial importância quando estes tumores têm localização retroperitoneal e na procura de metástases2. A tomografia de emissão de positrões (PET scan) poderá ter um papel na detecção da transformação maligna dos neurofibromas1.
A terapêutica destes tumores continua a ser sobretudo cirúrgica5. A ressecção cirúrgica radical com margens livres de doença é o procedimento de escolha1. Esta vai depender sobretudo da sua localização, na medida em que este procedimento é possível em 95% dos tumores nas extremidades, mas apenas em 20% dos que apresentam localização paraespinhal2. A RT é usada cada vez mais, dada a grande recorrência local destes tumores; no entanto, apesar de aumentar o tempo livre de doença, ainda é controverso se tem impacto significativo nas taxas de sobrevida a longo prazo2. Tal como a maioria dos sarcomas de tecidos moles, também os MPNST são tipicamente resistentes à QT. Apesar dos escassos dados publicados, a QT encontra-se geralmente indicada em crianças e jovens adultos com tumores com mais de 5 cm, irressecáveis ou com metástases na altura do diagnóstico1. Os tratamentos de primeira linha baseiam-se sobretudo em esquemas de doxorrubicina e ifosfamida, enquanto os de segunda linha, muito mais indefinidos, estão descritos com gemcitabina/docetaxel ou carboplatina/etoposido9.
Os MPNST, comparativamente aos restantes sarcomas de tecidos moles, têm uma taxa de recorrência local maior, ocorrendo em cerca de 20-40% após a ressecção cirúrgica10,11,12. Destes, 75% ocorrem nos primeiros dois anos de seguimento da doença12. Os principais factores de risco de recorrência local são a existência de margens de ressecção positivas e a sua localização (cabeça e pescoço)11. A metastização é frequente e é feita principalmente por via hematogénea13. Esta ocorre sobretudo para os pulmões e, por ordem decrescente de frequência, para os tecidos moles, osso, fígado, cavidade abdominal, glândulas suprarrenais, diafragma, mediastino, cérebro, ovários, rins e retroperitoneu4.
A sobrevida global aos 5 anos é pequena, variando desde os 12,8% aos 16%, dependendo do estudo em causa4. Os factores de mau prognóstico são: localização do tumor (cabeça, pescoço e tronco), a sua dimensão, diferenciação histológica, margens cirúrgicas positivas e a presença de NF11.
O caso clínico que descrevemos corresponde a uma situação muito rara de um grande tumor maligno da bainha dos nervos periféricos de localização torácica sem antecedentes de NF1 e sem ter realizado tratamentos prévios de RT. Além disso, a sua localização torácica torna-o uma entidade ainda mais rara. A sua apresentação clínica e idade de aparecimento é a descrita para este tipo de tumor, sendo o quadro de dor muito provavelmente secundário a compressão nervosa. O doente foi ubmetido a ressecção tumoral total com intuito life-saving, na medida em que o rápido rescimento da massa comprometia as estruturas vizinhas, condicionando dor e dispneia, e, desta forma, foi possível caracterizá-lo histologicamente. Uma vez que se trata de um tumor esporádico, sem antecedentes de NF1, a sua caracterização histológica foi difícil e demorada. Dada a sua grande dimensão, foi submetido a QT com o esquema habitual para os sarcomas e, posteriormente, RT. Finalmente, o prognóstico deste doente é mau, dado o subtipo histológico do sarcoma, a sua localização e a sua grande dimensão – sem dúvida o mais importante factor de prognóstico.
Bibliografia
1. Grobmyer S, Reith J, Shahlaee A, Bush C, Hochwald S. Malignant peripheral nerve sheath tumor: molecular pathogenesis and current management considerations. J Surg Oncol 2008; 97(4):340-349.
2. Gupta G, Maniker A. Malignant peripheral nerve sheath tumors. Neurosurg Focus 2007; 22(6):E12.
3. Shimizu J, et al. A case of intrathoracic giant malignant peripheral nerve sheath tumor in neurofibromatosis type I (von Recklinghausen’s disease). Ann Thorac Cardiovasc Surg 2008; 14(1):42-47.
4. Ducatman B, Scheithauer B, Piepgras D, Reiman H, Ilstrup D. Malignant peripheral nerve sheath tumors: a clinicopathologic study of 120 cases. Cancer 1986; 57: 2006-2021.
5. Strollo D, Rosado-de-Christenson M, Jett J. Primary mediastinal tumors part II: Tumors of the middle and posterior mediastinum. Chest 1997; 112(5):1344-1357.
6. Roviaro G, Montorsi M, Varoli F, Binda R, Cecchetto A. Primary pulmonary tumours of neurogenic origin. Thorax 1983; 38:942-945
7. Korf B. Malignancy in neurofibromatosis type 1. The Oncologist 2000; 5:477-485. [ Links ]
8. Gladish G, Sabloff B, Munden R, Truong M, Erasmus J, Chasen M. Primary thoracic sarcomas. Radio-Graphics 2002; 22:621-637.
9. Steins M, Serve H, Zulhsdorf M, et al. Carboplatin/ etoposide induces remission of metastasised malignant peripheral nerve sheath tumours (malignant schwannoma) refractory to first-line therapy. Oncol Rep 2002; 9:627-630.
10. Vauthey J, Woodruff J, Brennan M. Extremity malignant peripheral nerve sheath tumors (neurogenic sarcomas): A 10 -year experience. Ann Surg Oncol 1995; 2:126-131.
11. Anghileri M, Miceli R, Fiore M, et al. Malignant peripheral nerve sheath tumors: Prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006; 107:1065-1074.
12. Ramanathan R, Thomas J. Malignant peripheral nerve sheath tumours associated with von Recklinghausen’s neurofibromatosis. Eur J Surg Oncol 1999; 25:190-193.
13. Wanebo J, Malik J, Vandenberg S, Wanebo H, Driesen N, Persing J. Malignant peripheral nerve sheath tumors: a clinicopathologic study of 28 cases. Cancer 1993; 71(4):1247-1253.
Correspondência
e-mail:nelsonmarcal@gmail.com
Recebido para publicação/received for publication: 09.09.02
Aceite para publicação/accepted for publication: 09.10.08
