










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Pneumologia
versão impressa ISSN 0873-2159
Rev Port Pneumol v.16 n.2 Lisboa abr. 2010
Esclerose tuberosa com envolvimento pulmonar
Susana Ferreira1, Carla Nogueira1, Daniela Ferreira2, Sofia Neves2, Natália Taveira3
1Interna Complementar de Pneumologia
2Assistente Hospitalar de Pneumologia
3Assistente Hospitalar Graduada de Pneumologia CHVNG/Espinho EPE
Centro Hospitalar de Vila Nova de Gaia/Espinho EPE - Portugal
Directora: Dr.ª Bárbara Parente
Resumo
A esclerose tuberosa (ET) é uma doença rara, esporádica ou transmitida de forma autossómica dominante. Caracteriza-se pela tríade convulsões, atraso mental e angiofibromas faciais. O envolvimento pulmonar é raro e, quando ocorre, é mais frequente no sexo feminino. Os autores apresentam o caso de uma doente de 52 anos, não fumadora, com antecedentes conhecidos de epilepsia na infância e angiomiolipomas renais. Assintomática e sem alterações ao exame objectivo. Em tomografia do tórax realizada para avaliação da doença, foram detectadas formações microquísticas dispersas em ambos os campos pulmonares. Exame funcional respiratório normal. A ressonância magnética cerebral evidenciou tuberosidades corticais e nódulos subependimários calcificados. Concluiu–se pelo diagnóstico de ET (linfangioleiomiomatose, tuberosidades corticais, nódulos subependimários e angiomiolipomas renais). Os autores apresentam o caso pela raridade da doença e do envolvimento pulmonar, ainda que em fase assintomática.
Palavras-chave: Angiomiolipoma, esclerose tuberosa, linfangioleiomiomatose.
Tuberous sclerosis with pulmonary involvment
Abstract
Tuberous sclerosis (TS) is a rare, sporadic or autosomal dominant disease characterized by the triad of seizures, mental retardation and angiofibromas. Lungs are rarely involved in TS, and pulmonary involvement is almost always found in females. We report the case of a 52 year-old female, nonsmoker, with a history of seizures in childhood and renal angiomyolipomas. She presented no complaints and her physical exam was normal. Chest CT performed for the evaluation of the disease detected thin-walled pulmonary cysts in both lungs. Lung function tests were normal. Cortical tubers and calcified subependymal nodules were seen in cerebral magnetic resonance. Tuberous sclerosis was diagnosed (lymphangioleiomyomatosis, cortical tubers, calcified subependymal nodules and angiomyiolipomas). The authors present this case because of its rarity and the existence of pulmonary involvement, while still asymptomatic.
Key-words: Tuberous sclerosis, lymphangioleiomyomatosis, angiomyolipoma.
Introdução
A esclerose tuberosa (ET) foi reconhecida pela primeira vez em 1880 por Bourneville, que descreveu as manifestações neurológicas da doença e, daí, a denominação de doença de Bourneville1. A tríade característica da doença, convulsões, atraso mental e angiofibromas faciais, foi descrita em 1908 por Vogt2.
É uma doença rara, esporádica ou transmitida de forma autossómica dominante, multissistémica, que se caracteriza pelo crescimento de tumores benignos hamartomatosos localizados em múltiplos órgãos. Os órgãos mais frequentemente envolvidos são rim, cérebro, pele, pulmão e coração.
Foram identificadas duas alterações genéticas: uma mutação no braço longo do cromossoma 9 (conhecida como TSC1– tuberous sclerosis complex 1) e outra no braço curto do cromossoma 16 (conhecida como TSC2 – tuberous sclerosis complex 2), presentes em cerca de 80% dos casos3.
Afecta igualmente ambos os sexos, sem predisposição por raça, e pode surgir em qualquer idade, diferindo no entanto as manifestações da doença de acordo com a idade de apresentação. A incidência é de cerca de 1:60004.
O diagnóstico é clínico e baseado nos critérios revistos pela Tuberous Sclerosis Alliance (TS Alliance) e pelo National Institutes of Health (NIH) em 1998 (Quadro I)5.
Quadro I – Critérios de diagnóstico da esclerose tuberosa revistos pela Tuberous Sclerosis Alliance (TS Alliance) e pelo National Institutes of Health (NIH) em 1998
Considera-se definitivo o diagnóstico de ET na presença de dois critérios major ou um major e dois minor. O diagnóstico é provável na presença de um critério major e um minor. A ET é considerada possível na presença de um critério major ou dois ou mais critérios minor.
Relato de caso
Os autores apresentam o caso de uma doente de 52 anos, não fumadora, com antecedentes conhecidos de epilepsia na infância, pré-eclampsia e angiomiolipomas renais diagnosticados desde a adolescência, mantendo vigilância em consulta de urologia. Sem novas crises de epilepsia desde os 16 anos. Sem hábitos medicamentosos. Sem antecedentes familiares relevantes.
Referenciada à consulta de Pneumologia na sequência de alterações na TAC (tomografia axial computorizada) torácica, realizada para avaliação da doença, que evidenciou área em vidro despolido no lobo superior esquerdo e inúmeras formações microquísticas dispersas por ambos os campos pulmonares (Fig. 1). Assintomática e sem alterações ao exame objectivo.
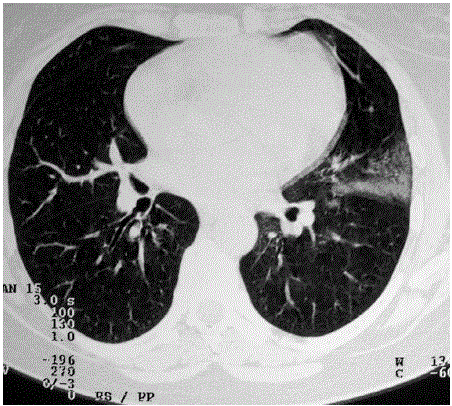
Fig. 1 –TAC (tomografi a axial computorizada) torácica evidenciando área em vidro despolido no lobo superior esquerdo e formações microquísticas dispersas pelos dois pulmões
Apresentava anemia normocítica normocrómica, velocidade de sedimentação de 15mm na 1.ª hora, marcadores víricos e serologias negativos, função hepática e renal e estudo imunológico sem alterações. Estudo funcional respiratório com FVC (forced vital capacity) de 2,61 L (95,4% prev); FEV1 (forced expiratory volume 1 second) de 2,44 L (105% prev); IT (índice de Tiffenau) de 93,5%; TLC (total lung capacity) 98,8%; VR (volume residual) 112,4%; VR/TLC 113,7%. Difusão do monóxido de carbono normal (92% prev). Gases do sangue arterial (FiO2 21%): pH 7,412; pO2 80,4; pCO2 34,5; HCO3 19,7; sO2 96,6%. Broncofibroscopia sem alterações, com microbiologia e citologias para células malignas negativas no lavado brônquico. Repete TAC abdominal, mantendo os angiomiolipomas renais já conhecidos (Fig. 2). Realizou ressonância magnética cerebral que demonstrou tuberosidades corticais, em localização frontal paramediana e temporal posterior direitas e nódulos subependimários calcificados nos ventrículos laterais (Fig. 3).
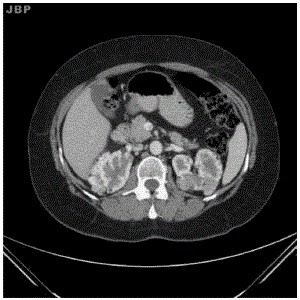
Fig. 2 – TAC (tomografi a axial computorizada) abdominal evidenciando angiomiolipomas renais bilaterais
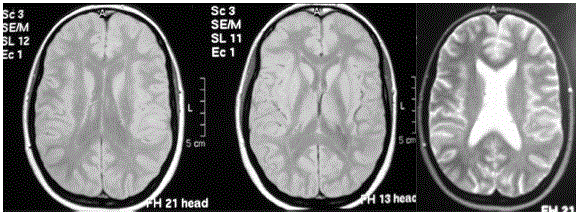
Fig. 3 – RM (ressonância magnética) cerebral - tuberosidades corticais e nódulos subependimários
Concluiu-se pelo diagnóstico de ET de acordo com as recomendações diagnósticas da TS Alliance e do NIH 3: linfangioleiomiomatose, tuberosidades corticais, nódulos subependimários e angiomiolipomas renais.
A doente mantém-se em vigilância e assintomática, sem novas alterações radiológicas.
Discussão
Na ET, as alterações cutâneas e neurológicas são as manifestações mais comuns, respectivamente 95 e 90% dos doentes5,6.
As manifestações neurológicas podem traduzir-se por convulsões, alterações psiquiátricas e comportamentais, alterações do desenvolvimento ou atraso mental e dependem essencialmente do tamanho e localização das tuberosidades corticais, nódulos su-bependimários e astrocitomas de células gigantes subependimários. As tuberosidades corticais não apresentam localização preferencial por nenhum lobo. Os nódulos subependimários encontram-se na parede lateral dos ventrículos e, em 5 -10% dos casos, podem degenerar em astrocitomas de células gigantes.
As manifestações cutâneas mais comuns são os angiofibromas faciais, máculas hipomelanocíticas, fibromas ungueais, periungueais ou gengivais e placas de shagreen (zonas de pele descolorada mais espessa e áspera no dorso, particularmente na região lombossagrada). Esta doente não apresentava nenhuma alteração cutânea.
As alterações renais são o terceiro achado clínico mais frequente e podem ocorrer como angiomiolipomas renais (>80% dos doentes com ET)7, quistos renais simples, rins poliquísticos e carcinoma de células renais. Os angiomiolipomas renais habitualmente são múliplos e bilaterais. A presença de sintomas, como dor, náuseas ou vómitos, está geralmente associada aos tumores com dimensões superiores a 4cm. O risco de hemorragia devido à formação de aneurismas também está aumentado nos tumores de maior diâmetro.
As alterações cardíacas manifestam-se em cerca de 50-60% dos doentes com ET, habitualmente durante a gestação ou no primeiro ano de vida, e habitualmente sob a forma de rabdomiomas, podendo causar sintomas essencialmente por obstrução ao fluxo ou por alterações do ritmo. No entanto, a maioria são assintomáticos e acabam por regredir espontaneamente nos primeiros anos de vida8,9.
As alterações oftalmológicas são referidas em 50 a 80% dos doentes, dependendo das séries7,10.
A alteração pulmonar mais frequente é a linfangioleiomiomatose (LAM). Surge quase exclusivamente no sexo feminino a partir da terceira ou quarta décadas de vida. O envolvimento pulmonar, previamente considerado uma manifestação rara, com uma prevalência inferior a 3% 11, pensa-se actualmente poder ter uma maior prevalência.
Um estudo recente que rastreou por TAC torácico mulheres com ET sem sintomas pulmonares demonstrou alterações císticas em cerca de 40%12.
Na LAM, as células musculares lisas sofrem uma proliferação anormal, comprometendo os bronquíolos, linfáticos e capilares. A elasticidade pulmonar vai reduzindo, com consequente diminuição da capacidade vital e aumento do volume residual. A LAM traduz-se pela presença de cistos nos pulmões, únicos ou múltiplos, variando de dimensões entre alguns milímetros até vários centímetros. A maior parte dos doentes manifesta-se com dispneia, pneumotórax, hemoptises ou, mais raramente, quilotórax.
A tríade clínica completa é incomum em doentes com envolvimento pulmonar, como aconteceu com esta doente.
No caso clínico apresentado não se observaram manifestações de todos os órgãos normalmente envolvidos na doença. Houve predomínio de envolvimento renal, neurológico e pulmonar.
A ET condiciona uma diminuição da sobrevida, sendo a doença renal e os tumores cerebrais as principais causas de morte. O prognóstico dos doentes com envolvimento pulmonar sintomático é reservado, sendo frequente a doença progressiva com insuficiência respiratória. No entanto, actualmente sabe-se que existem formas variadas da doença, algumas mais frustres e com um curso clínico mais favorável, como é o caso desta doente, até à data.
A ET deve ser encarada como uma doença crónica multissistémica, sendo os objectivos do tratamento a melhor qualidade de vida possível com o menor número de complicações e efeitos secundários.
Deve ser fornecido o tratamento de acordo com os sintomas, nomeadamente medicação anticonvulsiva e tratamento cirúrgico, se possível.
Até à data não existe nenhum tratamento eficaz para a LAM ou ET, à excepção do transplante pulmonar na fase terminal da doença. Devido à maior prevalência da LAM na mulher em idade reprodutiva, pensa-se que os estrogénios possam ter um papel na progressão da doença, pelo que alguns doentes recebem tratamento hormonal (progesterona), apesar de não ter sido demonstrado de forma conclusiva o seu benefício13,14.
Novas terapêuticas, como a rapamicina, apresentam já os primeiros resultados positivos. São necessários, no entanto, mais estudos para se confirmar eficácia e segurança15.
A resposta aos diferentes tratamentos testados é variável de doente para doente e nenhum tratamento demonstrou eficácia em todos os doentes com LAM.
Bibliografia
1. Bourneville DM. Scléreuse tubéreuse des circonvolutions cérébrales. Idiotie et épilepsie hémiplégiques. Bulletin de la Société médicale des hôpitaux de Paris 1884; 81-91.
2. Vogt H. Zur Diagnostik der tuberösen Sklerose. Zeitschrift für die Erforschung und Behandlung des jugendlichen Schwachsinns auf wissenschaftlicher Grundlage, Jena 1908; 2: 1-16.
3. Cheadle JP, Reeve MP, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Hum Genet 2000; 107(2):97-114.
4. Behrman RE, Kliegman RM, Jenson HB. In: Nelson, Tratado de pediatria. 17 ed. Rio de Janeiro: Elsevier; 2005; 589(2):2139.
5. Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998; 13(12):624-28.
6. Sogut A, Ozmen M, Sencer S, Calişkan M, Aydinli N, Ertuğrul T, et al. Clinical features of tuberous sclerosis cases. Turk J Pediatr 2002; 44(2):98 -101.
7. Smith HC, Watson GH, Patel RG, Super M. Cardiac rhabdomyomata in tuberous sclerosis: their course and diagnostic value. Arch Dis Child 1989; 64:196 -200.
8. Webb DW, Thomas RD, Osborne JP. Cardiac rhabdomyomas and their association with tuberous sclerosis. Arch Dis Child 1993; 68(3):367-370.
9. Bjornsson J, Henske EP, Bernestein J. Renal manifestations. In: Gomez MR, Sampson JR (Eds.). Tuberous sclerosis complex, 3th edition, Whittemore, New York: Oxford University Press1999, 181-193.
10. Kiribuchi K, Uchida Y, Fukuyama Y, et al. High incidence of fundus hamartomas and clinical significance of a fundus score in tuberous sclerosis. Brain Dev 1986; 8(5):509-17.
11. Castro M, Shepherd CW, Gomez MR, Lie JT, Ryu JH. Pulmonary tuberous sclerosis. Chest 1995; 107(1):189-195.
12. Moss J, Avila NA, Barnes PM, Litzenberger RA, Bechtle J, Brooks PG, et al. Prevalence and clinical characteristics of lymphangioleiomyomatosis in patients with tuberous sclerosis complex. Am J Respir Crit Care Med 2001; 164(4): 669-671.
13. Denoo X, Hermans G, Degives R, Foidart JM. Successful treatment of pulmonary lymphangioleiomyomatosis with progestins. Chest 1999; 115:276-279.
14. Chu SC, Horiba K, Usuki J, Avila NA, Chen CC, Travis WD, et al. Comprehensive evaluation of 35 patients with lymphangioleiomyomatosis. Chest 1999; 115:1041-1052.
15. Yates JR. Tuberous sclerosis. Eur J Hum Genet 2006; 14(10): 1065-1073. [ Links ]
Susana Alves Ferreira
Rua Conceição Fernandes
Vilar de Andorinho
4430-502 Vila Nova de Gaia, Portugal
Telefone: 00351912767155
e-mail: susalvesferreira@gmail.com
Recebido para publicação/received for publication:09.03.31
Aceite para publicação/accepted for publication:09.08.06
