










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Pneumologia
versão impressa ISSN 0873-2159
Rev Port Pneumol v.15 n.5 Lisboa out. 2009
Granulomatose de Wegener – Envolvimento otológico, nasal, laringotraqueal e pulmonar
Sandra Figueiredo 1
Laurentino Mendes Leal 2
António Morais 3
Adriana Magalhães 4
Teresa Oliveira 5
Venceslau Hespanhol 4
Carlos Dias 6
Gabriela Fernandes 3
Resumo
A granulomatose de Wegener é uma vasculite sistémica rara e idiopática caracterizada pelo atingimento dos pequenos vasos. A doença atinge, preferencialmente, as vias aérea superior e inferior e os rins, levando à formação de granulomas e necrose destes órgãos. As manifestações clínicas e o envolvimento orgânico variam largamente. O diagnóstico e tratamento precoce podem levar à recuperação total. No entanto, o atraso no diagnóstico pode ser fatal. Os autores apresentam o caso de uma doente de 33 anos com uma forma de apresentação grave e rara da doença, mas com uma evolução favorável após diagnóstico e tratamento adequado.
Palavras-chave: Granulomatose de Wegener, forma limitada grave.
Wegener granulomatosis – Otologic, nasal, tracheobronchial and pulmonary involvement
Abstract
Wegener granulomatosis is a rare systemic idiopathic disease characterized by involvement of small vessels – medium and small arteries, venules, arterioles and ocasionally large arteries. This disease has predilection for the upper and lower respiratory tract and the kidney, with granulomatous inflamation and necrosis. Clinical manifestations and organ involvement of the disease vary widely. Early diagnosis and treatment may lead to a full recovery. Without treatment, Wegeners granulomatosis can be fatal. The authors present a case of a 33 year-old female, with severe disease, but with good outcome, after adequate diagnosis and treatment.
Key-words: Wegener´s granulomatosis, severe limited form.
Introdução
Em 1936, o patologista alemão Friedrich Wegener1 publicou um caso clínico de morte por doença das vias aéreas superiores e insuficiência respiratória. Em 1939, reportou a primeira série de casos da doença que ficaria conhecida como granulomatose de Wegener (GW). Fiemberg2, em 1953, introduziu, conjuntamente com Carrington e Liebow3, o conceito de granulomatose de Wegener limitada, em que a doença poderia restringir-se a apenas um ou mais órgãos e, não necessariamente, apresentar envolvimento das vias aéreas superiores, inferiores e rins. Este conceito foi reforçado por Cassan e colaboradores4.
A GW é uma doença sistémica rara e idiopática que atinge, preferencialmente, os pequenos vasos (pequeno e médio calibre) – vénulas e arteríolas. Ocasionalmente, pode comprometer artérias de grande calibre.
Caracteriza-se por inflamação granulomatosa dos vasos das vias aéreas superiores, inferiores e rins, com vasculite e necrose.
Segundo estudos americanos, a prevalência é de 3 casos/100 000 indivíduos. A maioria dos doentes apresenta-se entre os 20 e os 40 anos5. Os homens parecem ser os mais susceptíveis, com uma razão homem/mulher de 1,5:1,0. A doença é rara entre os indivíduos de raça negra.
A forma clássica caracteriza-se por inflamação granulomatosa das vias aéreas superiores, inferiores e rins. A forma limitada manifesta-se, apenas, nas vias aéreas superiores ou pulmão. Esta classificação é, no entanto, controversa, pois a biópsia renal em doentes assintomáticos pode revelar atingimento renal em 80% dos casos6.
A remissão completa pode ser obtida em 75% dos casos. Se não for tratada, a GW tem uma mortalidade de 80% no primeiro ano. A sobrevivência dos doentes aumentou significativamente nos últimos anos devido à utilização de terapêuticas imunossupressoras7.
Antes do aparecimento dos corticosteróides, a granulomatose de Wegener (GW) clássica era uma doença fatal, com uma sobrevida média muito baixa. Mais tarde, em 1973, a combinação de corticosteróides com ciclofosfamida revelou obter remissões sustentadas e sobrevida mais prolongada8.
Os sintomas gerais são febre, anorexia, emagrecimento, fadiga e fraqueza, além dos sintomas relacionados com o órgão atingido7.
A maioria dos doentes tem envolvimento do tracto respiratório superior e/ou inferior. Cerca de 90% das queixas envolvem o nariz e os seios perinasais: obstrução nasal crónica com rinorreia mucopurulenta, ulceração e edema da mucosa nasal, parosmia, epistáxis e cefaleias. A deformidade do nariz em sela, devido à destruição da cartilagem nasal, ocorre numa pequena percentagem dos casos9. Estas lesões aumentam a susceptibilidade a infecções, pelo que o diagnóstico diferencial pode ser dificultado. O envolvimento otológico é de cerca de 35%10 e relaciona-se com a obstrução da tuba auditiva por atingimento da rinofaringe. Os sintomas mais frequentes são otite média, com perfuração da membrana timpânica, otalgia e otorreia10. O envolvimento pulmonar é comum (70 a 90% dos casos), sendo as queixas principais tosse produtiva, dispneia, hemoptise, dor e desconforto torácico. Na maioria dos casos, os sintomas respiratórios são acompanhados por alterações radiológicas, nomeadamente opacidades nodulares, por vezes cavitadas, com dimensões variáveis, múltiplas e bilaterais11,12. O envolvimento laringotraqueal pode ser assintomático, mas sintomas, como dispneia de esforço, estridor, hemoptises e rouquidão, são comuns13.
A estenose subglótica é descrita em cerca de 23% dos doentes, podendo em 2,3% ser o sintoma de apresentação14. A glomerulonefrite focal necrotisante é a lesão mais comum e pode associar-se a insuficiência renal. No sedimento urinário podem estar presentes hematúria e leucocitúria15. O atingimento cutâneo, ocular e articular, também pode ocorrer, mas é menos frequente.
O achado histológico mais importante inclui a presença de necrose, vasculite e inflamação granulomatosa. A necrose tende a ser extensa. As lesões granulomatosas apresentam polimorfonucleares, células plasmáticas, raros eosinófilos e linfócitos e, especialmente, células gigantes multinucleadas. A vasculite é caracterizada pelo envolvimento de arteríolas e vénulas, com consequente obstrução do lúmen vascular16.
Caso clínico
Doente de 33 anos, sexo feminino, raça caucasiana, sem antecedentes pessoais relevantes, referenciada à consulta de ORL por quadro de otalgia e hipoacusia à direita, tendo sido diagnosticada otite média com efusão, sujeita a miringotomia e medicada com antibioterapia, sem melhoria significativa, ficando em vigilância na consulta de ORL.
Além das queixas localizadas ao ouvido direito, iniciou queixas da nasofaringe com aparecimento de úlceras faríngeas e nasais, cujas biópsias foram inconclusivas. Progressivamente, iniciou quadro de artralgias de predomínio nos membros inferiores, astenia e mal-estar geral, além de úlceras cutâneas, tendo sido internada para estudo após o início da sintomatologia. Apresentava-se pálida, emagrecida e apirética, com várias lesões ulcerativas da pele. Não apresentava outras alterações no exame físico, nomeadamente na auscultação pulmonar. O exame otorrinolaringológico apresentava mucosa nasal íntegra, com algumas crostas e escassas úlceras na orofaringe. O exame anatomopatológico das biópsias realizadas às úlceras cutâneas e valvulares revelou um processo ulcerativo sugestivo de vasculite sistémica, salientando-se a presença de lesões vasculares de vasos de pequeno calibre de tipo venoso, na ausência de lesões granulomatosas.
As biópsias da nasofaringe foram inconclusivas por serem constituídas predominantemente por material necrótico. Analiticamente, apresentava anemia hipocrómica (Hb – 10 mg/dl), proteína C reactiva e VS elevada (289 mg/L e 110 mm 1ª hora, respectivamente).
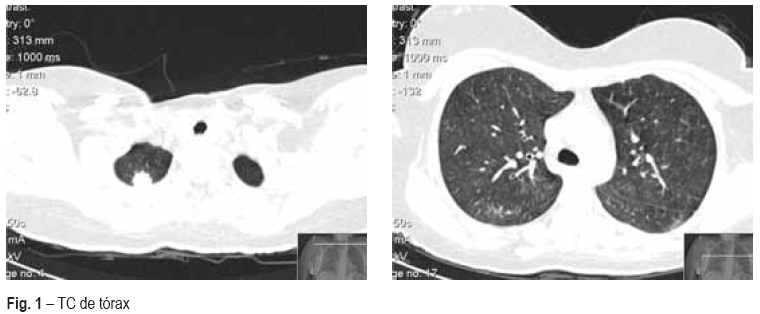
A função renal estava preservada. O exame de urina não demostrava sedimento activo. A titulação do ANCA PR3 era positivo (99U/ml). A tomografia computorizada (TC) do torax demonstrava a presença de dois nódulos pulmonares à direita, com imagem em vidro despolido no lobo superior direito (Fig. 1). A endoscopia digestiva alta mostrava úlceras orofaríngeas, sem outras alterações, e a broncofibroscopia não mostrava alterações significativas, além de mucosa friável. No exame cultural da expectoração isolaram-se os seguintes microrganismos: Pseudomonas aeruginosa, Staphylococcus aureus resistentes à meticilina (SAMR) e Candida albicans. A situação clínica da doente evoluiu desfavoravelmente, apesar da antibioterapia de largo espectro instituída (vancomicina, piperacilina/tazobactan e fluconazol). Doze dias após o internamento, a doente desenvolveu insuficiência respiratória com falência respiratória, pelo que foi entubada e admitida em unidade de cuidados intensivos (UCI). Embora sem diagnóstico histológico confirmado, iniciou pulsos de metilprednisolona e ciclofosfamida.
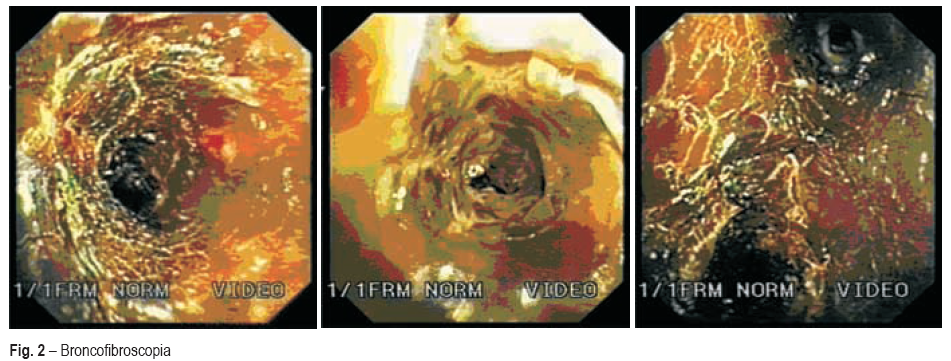
A broncofibroscopia realizada nessa altura evidenciava mucosa recoberta, em toda a extensão, por película translúcida e acastanhada, parecendo necrose generalizada (Fig. 2). Ainda na UCI, realizou biopsia pulmonar cirúrgica das lesões pulmonares descritas, confirmando tratar-se de granulomatose de Wegener. Já com terapêutica instituída, ciclofosfamida e metilprednisolona, a doente melhorou progressivamente, tendo tido alta da UCI após 15 dias de internamento e do hospital após 70 dias.
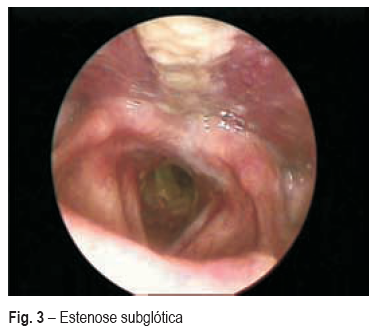
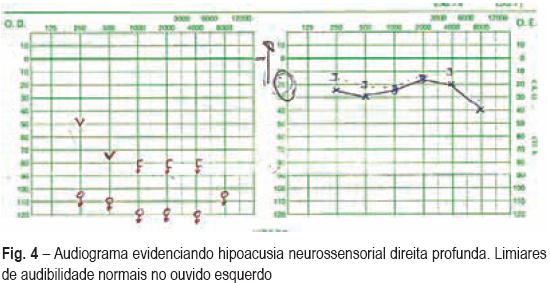
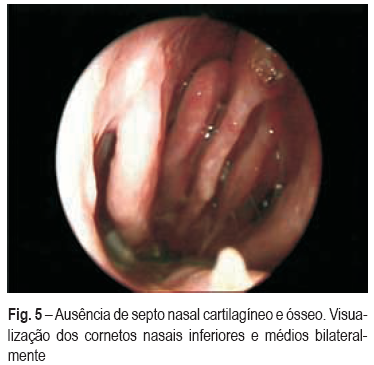
Um mês após a alta, recorreu ao SU por dificuldade respiratória e estridor, tendo sido demonstrada estenose subglótica (Fig. 3) a cerca de 2 cm abaixo das cordas vocais, com cerca de 6 cm de extensão, condicionando obstrução do lúmen traqueal em cerca de 80%, com necessidade de dilatação mecânica e colocação de prótese traqueal. As sequelas otorrinolaringológicas foram a destruição completa do septo nasal (Fig. 4) e surdez à direita (Fig. 5). Encontra-se em seguimento em consulta de ORL, pneumologia (vigilância clínica e endoscópica da estenose e da prótese) e medicina interna (doenças autoimunes).
Discussão
A granulomatose de Wegener é uma vasculite granulomatosa, cuja etiologia é desconhecida, provavelmente mediada imunologicamente.
Os eventos inflamatórios têm uma afinidade invulgar para o epitélio respiratório ciliado da cavidade nasal, seios perinasais e árvore traqueobrônquica. As manifestações otorrinolaringológicas são frequentes.
A estenose traqueal é menos comum, podendo surgir como manifestação inicial, numa fase avançada ou até de resolução da doença e mesmo sob terapia imunossupressora17.
Os sintomas podem variar de tosse e dispneia de esforço a estridor com compromisso da via aérea13. Pode ser suspeitada na tomografia computorizada da traqueia (reconstrução tridimensional) e na espirometria (achatamento do ramo inspiratório e expiratório da curva débito/volume) e confirmada por broncofibroscopia A estenose subglótica ou da traqueia proximal com cicatrização circunferencial e obstrução luminal pode surgir entre 10 a 16%18 e em algumas séries até 26%14 dos casos, em qualquer momento da evolução da doença. A causa poderá ser o próprio processo inflamatório imunomediado, embora não se possa excluir a hipótese de complicação de entubação orotraqueal ou infecção das vias aéreas superiores, sendo o diagnóstico de certeza apenas fornecido por estudo anatomopatológico17.
O tratamento pode implicar dilatação mecânica, infiltrações intralesionais com corticóide, colocação de prótese ou até tratamento cirúrgico17,18.
O diagnóstico de granulomatose de Wegener torna-se um verdadeiro desafio para o médico.
A hipótese deve ser colocada perante sintomas relacionados com atingimento das vias aéreas superiores, inferiores e do rim. O diagnóstico deve ser estabelecido logo que possível e o tratamento instituído com a maior brevidade, de modo a permitir a remissão, reduzindo a morbilidade e a mortalidade da doença.
Neste caso, apesar da suspeita clínica, as biopsias do tecido nasal e da nasofaringe foram inconclusivas e a ausência de atingimento renal afastaram, de inicío, a possibilidade de granulomatose de Wegener. No entanto, perante a positividade do anticorpo anticitoplasma do neutrófilo contra o antigénio proteinase 3 (ANCA-PR3) e o agravamento clínico (necessidade de entubação e ventilação invasiva), foi decidido o início de tratamento imunossupressor, com resposta favorável.
Cerca de 90% dos doentes têm ANCA-c positivos, a maioria produzindo um padrão com especificidade PR3 (proteinase 3) e menos de 5% com especificidade MPO (mieloperoxidase).
A sensibilidade e especifidade da titulação destes autoanticorpos é maior para forma clássica da doença. Cerca de 40% dos doentes com forma limitada têm ANCA negativos, pelo que a ausência destes não exclui a doença19. Por este motivo, os novos critérios da Chapel Hill Consensus Conference Nomenclature, de 1994, não incluem os ANCA como critério de diagnóstico20.
O tratamento instituído consistiu na administração de pulsos de metilprednisolona (equivalente a 1 mg/kg/dia prednisolona) e ciclofosfamida (1g/dia). As recomendações para o tratamento da granulomatose de Wegener21 privilegiam o uso combinado de prednisolona (ou equivalente) e ciclofosfamida, especialmente nos casos com compromisso vital, ou seja, elevação dos valores de creatinina superiores a 2mg/dl, envolvimento pulmonar com hipoxemia, atingimento do sistema nervoso central e perfuração/isquemia intestinal. As doses recomendadas de são de 1,5-2 mg/kg/dia, sendo a leucopenia um efeito colateral previsível.
Esta doente efectuou um esquema de ciclofosfamida 1g/dia devido ao desenvolvimento de neutropenia iatrogénica, com necessidade de administração de factores de crescimento.
Relativamente à corticoterapia, a dose recomendada é de 1mg/dia (com um máximo de 60-80 mg) de prednisolona oral (ou equivalente), com o objectivo de reduzir a inflamação até ao início da acção da ciclofosfamida (estimado em cerca de 7 a 14 dias). Vários esquemas têm sido utilizados, mas em geral o mais utilizado é o uso inicial de doses elevadas durante 2 a 4 semanas. A redução poderá ser iniciada com o início de melhoria clínica até um basal de 20 mg/dia, durante cerca de 6 a 9 meses. O uso de metilprednisolona nos 3 primeiros dias poderá estar indicado quando existe atingimento renal grave e respiratório como forma de apresentação.
Novos estudos têm recomendado a substituição de ciclofosfamida por metrotrexato. No tratamento inicial, o metotrexato pode ser usado em monoterapia ou associado à corticoterapia em doentes sem compromisso orgânico vital. No entanto, ainda está por demonstrar a eficácia a longo prazo deste fármaco22.
Uma vez atingida a fase de remissão, estes doentes devem permanecer sob vigilância, devendo estar sempre presente a possibilidade de recaída23,24.
O advento de novos imunossupressores como agentes biológicos (ex. infliximab) parecem promissores no tratamento de vasculites, estando ainda por comprovar a sua eficácia e segurança22.
Este caso demonstra, pela gravidade e morbilidade que acarretou, que, ao contrário do que está descrito na literatura, a forma limitada da GW pode ter uma evolução mais agressiva do que a esperada.
Bibliografia
1. Wegener F. Uber generalisierte, septische Gefässerkrankungen. Verh Dtsch Ges Pathol 1936; 29:202-210. [ Links ]
2. Fienberg R. Necrotizing granulomatosis and angiitis of the lungs. Am J Clin Pathol 1953;23:413-28.
3. Carrington CB, Liebow M. Limited forms of angiitis and granulomatosis of Wegeners type. Am J Med 1966; 41:497-527.
4. Cassan SM, Coles DT, Harisson EG. The concept of limited forms of Wegeners granulomatosis. Am J Med 1970 ;49:366-379.
5. Koldingsnes W, Nossent H. Epidemiology of Wegeners granulomatosis in Nothern Norway. Art Rheum 2000; 43(11):2481-2487.
6. Duna GF, Galperin C, Hoffman GS. Wegeners granulomatosis. Rheum Dis Clin North Am 1995; 21:949-986.
7. Fauci AS, Haynes BF, Katz P, Wolff SM. Wegeners granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 1983; 98(1):76-85.
8. Fauci AS, Wolf SM. Wegener´s granulomatosis: studies in eighteen patients and a review of the literature. Medicine (Baltimore) 1973; 52(6):535-561.
9. Jones NS. Nasal manifestations of rheumatic diseases. Ann Rheum Dis 1999; 58:589-590.
10. Hartl DM, Aidan P, Brugière O, Sterkers O. Wegeners granulomatosis presenting as a recurrence of chronic otitis media. Am J Otol 1998;19(1):54-60.
11. Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegeners granulomatosis. A clinical and imaging study of 77 cases. Chest 1990; 97:906-912.
12. Lee SK, Kim TS, Fujimoto K, et al. Thoracic manifestations of Wegener´s granulomatosis: CT findings in 30 patients. Eur Radiol 2003; 13:43-51.
13. Daum TE, Specks U, Colby TV, Edell ES, Brutinel MW, Prakash UB, DeRemee RA. Tracheobronchial involvement in Wegeners granulomatosis Am J Respir Crit Care Med 151:522-526.
14. Langford CA, Sneller MC, Hallahan CW, Hoffman GS, Kammerer WA, Talar-Williams C, Fauci AS, Lebovics RS. Clinical features and therapeutic management of subglottic stenosis in patients with Wegeners granulomatosis. Arthritis Rheum 1996; 39(10):1754-1760.
15. Seo P, Stone JH. The antineutrophil cytoplasmic antibody-associated vasculitides. Am J Med 2004; 117: 39.
16. Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegeners granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315-333.
17. Lebovics RS, Hoffman GS, Leavitt RY, Kerr GS, Travis WD, Kammere W, Hallahan C, Rottem M, Fauci AS. The management of subglottic stenosis in patients with Wegeners granulomatosis. Laryngoscope 1992; 102(12 Pt 1):1341-1345.
18. Solans-Laqué, et al. Clinical features and therapeutic management of subglottic stenosis in patients. Lupus 2008; 17:832-836.
19. Rao JK, Allen NB, Feussner JR, Weinberger M. A prospective study of antineutrophil cytoplasmic antibody (c-ANCA) and clinical criteria in diagnosing Wegeners granulomatosis. Lancet 1995 7; 346(8980):926-931.
20. Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitis: the proposal of an international consensus conference. Arthritis Rheum 1994; 37:187-192.
21. Hoffman GS. Wegener´s granulomatosis. Curr Opin Reumathol 1993; 5(1):11-17.
22. D. Jayne. Evidence-based treatment of systemic vasculitis. Rheumatology 2000; 39:585-595.
23. Sanders J-S F, Slot MC, Stegeman CA, Jayne D, Rasmussen N. Maintenance Therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:2072-2073.
24. Sanders J-SF, Huitma MG, Kallenberg CGM, Stegeman CA. Prediction of relapses in PR3-ANCA–associated vasculitis by assessing responses of ANCA titres to treatment. Rheumatology 2006; 45(6):724-729.
1 Internato Médico. Serviço de Pneumologia
2 Internato Médico. Serviço de Otorrinolaringologia
3 Assistente Hospitalar. Serviço de Pneumologia
4 Assistente Hospitalar Graduado. Serviço de Pneumologia
5 Assistente Hospitalar Graduada. Unidade de Cuidados Intensivos Polivalentes
6 Assistente Hospitalar Graduado. Serviço de Medicina Interna
Hospital de São João (EPE), Porto
E-mail: sandrabugalho@iol.pt
Recebido para publicação/received for publication: 08.12.22
Aceite para publicação/accepted for publication: 09.03.05
