










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkJornal Português de Gastrenterologia
versão impressa ISSN 0872-8178
J Port Gastrenterol. vol.19 no.6 Lisboa nov. 2012
https://doi.org/10.1016/j.jpg.2012.04.033
Cirrose hepática em doente jovem - caso clínico e revisão da literatura
Hepatic cirrhosis in young patient - case report and literature review
Ana Catarina Lagos∗, Inês Marques, Beatriz Rodrigues, Jorge Reis e Beatriz Neves
Serviço de Gastrenterologia II, Hospital Pulido Valente (CHLN), Lisboa, Portugal
*Autor para correspondência
Resumo
Os autores reportam o caso de um doente de 25 anos de idade, com um quadro clínico compatível com pneumonia que condicionou o primeiro episódio de descompensação de doença hepática crónica, sem diagnóstico prévio. Os exames complementares de diagnóstico revelaram a presença de Doença de Wilson.
A Doença de Wilson é uma entidade rara, autossómica recessiva, em que ocorre uma diminuição da excreção biliar de cobre, resultando na sua acumulação no fígado, cérebro, rim e córnea. A apresentação clínica desta entidade inclui um espectro alargado de manifestações, entre as quais se inclui a doença hepática crónica descompensada, como ocorreu no caso clínico descrito.
PALAVRAS-CHAVE Doença de Wilson; Doença hepática crónica descompensada
Abstract
The authors present the case of 25-year-old male with pneumonia, that caused the first decompensation of chronic liver disease, without previous diagnosis. Diagnostic workup revealed the presence of Wilson disease.
Wilson disease is a rare, autosomal recessive disorder in which inadequate hepatic copper excretion leads to its accumulation in the liver, brain, kidney and cornea. The clinical presentation include a broad spectrum of symptoms, like decompensated chronic liver disease, as in our case.
KEYWORDS Wilson disease; Decompensated chronic liver disease
Introdução
A Doença de Wilson (DW), descrita pela primeira vez em 1912 por Kinnear Wilson1, é uma doença rara, hereditária, de transmissão autossómica recessiva, caracterizada por acumulação de cobre no fígado, cérebro, rins e córnea2. A prevalência da DW é de cerca de 1:30 000 e a idade de apresentação varia entre os 3 e os 55 anos de idade3. Em Portugal, estima-se que, entre 2005 e 2008, tenham surgido 10 novos casos, conforme registo na base de dados internacional «eurowilson». As manifestações clínicas da DW podem atingir múltiplos órgãos e são extremamente variáveis, pelo que é necessário um elevado índice de suspeição para o seu diagnóstico.
Os autores apresentam um caso clínico de DW num adulto jovem, cuja primeira manifestação foi sob a forma de doença hepática crónica descompensada, sem diagnóstico prévio.
Caso clínico
Doente do sexo masculino de 25 anos de idade, sem antecedentes pessoais relevantes, nomeadamente hábitos alcoólicos ou toxicofílicos.
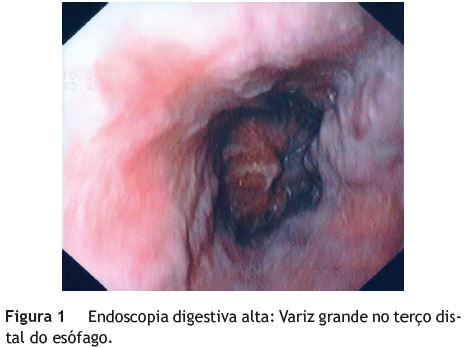
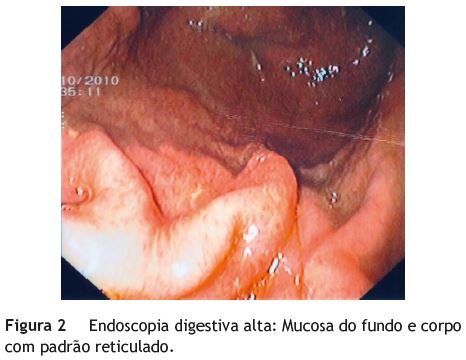
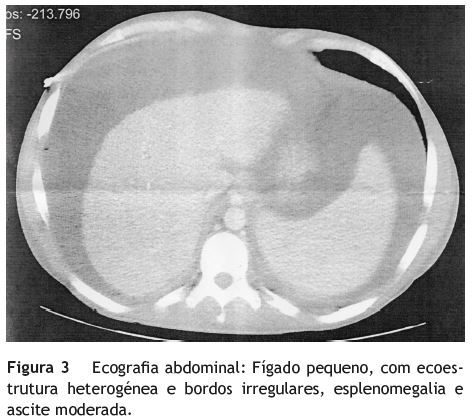
O doente recorreu ao Serviço de Urgência por um quadro clínico de febre e tosse com expetoração mucopurulenta com uma semana de evolução. À entrada encontrava-se febril, hemodinamicamente estável e apresentava diminuição do murmúrio vesicular na base do hemitórax direito. Analiticamente salientava-se aumento dos parâmetros inflamatórios, trombocitopenia (plaquetas de 65 000), prolongamento do tempo de protrombina com INR de 1,94, AST:116 U/L (valor de referencia (v.ref): 15-39 U/L), ALT: 96 U/L (v.ref: 8-37 U/L), bilirrubina total: 1,3 mg/dL (v.ref: 0-1 mg/dL), albumina:1,6 mg/dL (v.ref: 3,4-5,0 mg/dL) e função renal sem alterações. Na radiografia de tórax apresentava condensação na base do hemitórax direito. O doente foi internado no Serviço de Pneumologia com a hipótese diagnóstica de pneumonia hipoxemiante. Iniciou antibioterapia empírica e houve necessidade de ventilação não invasiva por insuficiência respiratória parcial, com melhoria do quadro. Durante o internamento, por apresentar epigastralgias e vómitos, realizou endoscopia digestiva alta, que revelou no terço distal do esófago, variz grande sem manchas vermelhas ou ponto de rotura (fig. 1) e mucosa do fundo e corpo com padrão em mosaico (fig. 2). Paralelamente, verificou-se agravamento clínico com aumento do volume abdominal e edema marcado dos membros inferiores. Realizou ecografia abdominal, que revelou fígado pequeno de ecoestrutura heterogénea, compatível com cirrose, esplenomegalia de 17 cm e ascite em moderada quantidade (fig. 3). O doente foi transferido para o nosso Serviço de Gastrenterologia com a hipótese diagnóstica de doença hepática crónica (DHC) descompensada (Child Pugh C; MELD 14), tendo sido submetido a paracentese diagnóstica, que excluiu peritonite bacteriana espontânea e revelou um gradiente de albumina soro-ascite > 1,1 g/dL, compatível com hipertensão portal. Da investigação etiológica do quadro de DHC, salientavase serologias negativas para os vírus da hepatite A, B, C, citomegalovírus, Epstein-Barr, Herpes simplex 1 e 2 e imunodeficiência humana adquirida 1 e 2, cinética do ferro e função tiroideia sem alterações, alfa 1 antitripsina e alfa fetoproteína dentro dos valores de referência, autoimunidade (ANA, anti-DNA, AMA, ASMA, anti-LKM) negativa e imunoglobulinas sem alterações. Devido à idade e sintomatologia do doente, foi também doseado o nível sérico da ceruloplasmina, que se revelou francamente baixo 3 mg/dL (v.ref: 22-58 mg/dl) e o doseamento urinário de cobre em 24 horas, que se encontrava aumentado 4,4_mol (v.ref < 0,78_mol). Os valores obtidos foram compatíveis com DW. O doente foi observado pela Oftalmologia, que confirmou a presença dos anéis de Kayser-Fleischer. Foi também observado pela Neurologia, que excluiu alterações no exame neurológico e realizou ressonância magnética craneo-encefálica, que não revelou alterações. O doente iniciou tratamento com trientina 250 mg 3xdia, acetato de zinco 50 mg 3xdia e diuréticos. Efetuou também laqueação elástica da variz esofágica. Verificou-se melhoria progressiva do quadro clínico-laboratorial.
O doente teve alta assintomático (Child Pugh B; MELD 7), referenciado para a consulta de Hepatologia, onde efetuou o estudo genético que revelou heterozigotia composta para as mutações c.3402delC e c.3694A>C. Foi efetuado o rastreio aos familiares de primeiro grau, nomeadamente à mãe do doente, que não apresentou mutações, ao irmão mais velho, que revelou ser portador heterozigótico para a mutação c.3402delC e ao irmão mais novo, que revelou ser portador heterozigótico para a mutação c.3694A>C. Não foi possível efetuar o rastreio ao pai do doente, uma vez que faleceu por neoplasia do pulmão aos 40 anos. De salientar que ambos os irmãos não apresentavam clínica sugestiva de DW.
O doente já cumpriu um ano de follow-up na consulta de Hepatologia, encontrando-se assintomático.
Discussão
A DW caracteriza-se pela excreção biliar inapropriada de cobre, resultando na acumulação deste metal no fígado, cérebro, rins e córnea. A alteração na excreção de cobre resulta de mutações no gene ATP7B (proteína transportadora do cobre) que se localiza no cromossoma 13. Atualmente, estão descritas mais de 500 mutações neste gene, sendo a mais frequente a His. Salienta-se que na família do nosso doente não foi identificada esta mutação. A multiplicidade de mutações identificadas até ao momento pode tornar o diagnóstico genético complexo, sendo a maioria dos doentes heterozigótico composto, como no caso apresentado.
A expressão clínica da DW é muito variável, manifestando-se geralmente através de doença hepática ou neuropsiquiátrica. A doença hepática é mais frequente no jovem3, tal como no caso clínico descrito, podendo apresentar-se de forma assintomática, por alterações das provas hepáticas, esteatose, hepatite aguda, doença hepática crónica ou mesmo falência hepatica4. A apresentação neurológica surge habitualmente na 2.a ou 3.a década de vida5 e compreende os sintomas parkinsónicos e os pseudo-bulbares, nomeadamente disfagia e disartria. A apresentação psiquiátrica, sem outros sintomas associados, ocorre em cerca de 20% dos doentes, sendo a depressão a sintomatologia mais frequente6. Os anéis de Kayser-Fleischer resultam da deposição de cobre na membrana de Descemet da córnea, podem ser observados através da lâmpada de fenda e estão presentes em 50-60% dos doentes com envolvimento hepático e em 90% dos doentes com doença neurológica7.
Na doença hepática de etiologia não conhecida, quando o doente apresenta ceruloplasmina < 20 mg/dL, cuprúria > 0,6_mol/24 h e anéis de Kayser-Fleischer, pode-se estabelecer o diagnóstico de DW, sem ser necessário recorrer a outros exames complementares de diagnóstico8, tal como se verificou no nosso caso clínico. Quando estes 3 critérios não estão presentes, torna-se necessário realizar biópsia hepática para quantificar o cobre hepático.
A terapêutica farmacológica é a pedra angular no tratamento destes doentes, tendo maior eficácia se iniciada precocemente e mantida ao longo da vida. As armas terapêuticas disponíveis são os quelantes do cobre, nomeadamente a penicilamina e a trientina, e o acetato de zinco, que diminui a absorção intestinal de cobre, podendo ser utilizado em associação com os quelantes. No caso do nosso doente, optou-se por utilizar a trientina, uma vez que este fármaco, quando administrado em doses apropriadas, tem eficácia semelhante à penicilamina e tem a vantagem de apresentar menos efeitos adversos8.
Os familiares em primeiro grau de um doente com DW devem efetuar o rastreio da doença.
A DW quando diagnosticada e tratada atempadamente, geralmente apresenta bom prognóstico, não estando preconizado o rastreio para o carcinoma hepatocelular3.
Bibliografia
1. Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain. 1912;34:295-507. [ Links ]
2. Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327-37. [ Links ]
3. Demetri GD. Gastrointestinal Stromal Tumours. Em: Sleisenger and Fordtrans Gastrointestinal and Liver Disease: Pathophysiology/ Diagnosis/Management. Filadélfia, PA: Saunders Elsevier; 2010. p. 1249-57. [ Links ]
4. Ferlan-Marolt V, Stepec S. Fulminant Wilsonian hepatitis unmasked by disease progression: report of a case and review of the literature. Dig Dis Sci. 1999;44:1054-8. [ Links ]
5. Oder W, Grimm G, Kollegger H, Ferenci P, Schneider B, Deecke L. Neurological and neuropsychiatric spectrum of Wilsons disease. A prospective study in 45 cases. J Neurol. 1991;238: 281-7. [ Links ]
6. Dening TR, Berrios GE. Wilsons disease; Psychiatric symptoms in 195 cases. Arch Gen Psychiatry. 1989;46:1126. [ Links ]
7. Tauber J, Steinert RF. Pseudo-Kayser-Fleischer ring of the córnea associated with non-Wilsonian liver disease. A case report and literature review. Cornea. 1993;12:74-7. [ Links ]
8. Roberts E, Schilsky M. Diagnosis and treatment of Wilson disease: an update. AASLD Practice Guidelines. 2008:2089-105. [ Links ]
Conflito de interesses
Os autores declaram não haver conflito de interesses.
*Autor para correspondência
Correio eletrónico: catarina.lagos@gmail.com (A.C. Lagos).
Recebido a 30 de julho de 2011; aceite a 14 de dezembro de 2011

{kind=link}