










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkJornal Português de Gastrenterologia
versão impressa ISSN 0872-8178
J Port Gastrenterol. vol.19 no.1 Lisboa jan. 2012
Pancreatite autoimune e diagnóstico diferencial com a neoplasia do pâncreas: A propósito de um caso clínico.
Juliana Oliveira1*, Gorete Jorge2, António Milheiro2, Francisco Castro Sousa3
1Interna do Internato Complementar de Cirurgia Geral
2Assistente Graduado
3Professor Catedrático de Cirurgia da FMUC, Director da Clínica Universitária de Cirurgia III Serviço de Cirurgia III dos Hospitais da Universidade de Coimbra
*Autor para correspondência
RESUMO
A Pancreatite Auto-imune (PAI) é um tipo de pancreatite crónica caracterizada, histológicamente, por um infiltrado linfoplasmocitário e fibrose periductal. O quadro clínico pode mimetizar um carcinoma da cabeça do pâncreas, principalmente nos casos de doença cefálica, sendo caracterizado por icterícia obstructiva, dor abdominal e perda ponderal. Esta entidade clínica pode estar associada a outras doenças auto-imunes e o diagnóstico tem por base os critérios diagnósticos sugeridos pela Clínica Mayo e pela Sociedade Japonesa do Pâncreas, em que associam as características histológicas e radiológicas à elevação da IgG4 e à resposta ao tratamento com corticóides.
Apresentamos um caso clínico de uma doente submetida a Duodenopancreatectomia cefálica, devido a um diagnóstico pré-operatório (clínico e radiológico) de carcinoma da cabeça do pâncreas. O estudo anatomo-patológico revelou tratar-se de uma pancreatite linfoplasmocitária e o estudo imunológico posterior corroborou o diagnóstico de Pancreatite Auto Imune.
Este artigo tem como objectivo alertar para o diagnóstico diferencial com o carcinoma da cabeça do pâncreas; de modo a evitar a realização de uma intervenção com taxas de morbilidade e mortalidade relevantes numa entidade nosológica sensível à terapêutica com corticóides.
PALAVRAS-CHAVE: Pancreatite autoimune, carcinoma da cabeça do pâncreas, icterícia obstructiva, infiltrado linfoplasmocitário, IgG4, duodenopancreatectomia cefálica.
Autoimmune pancreatitis and differential diagnosis with neoplasm of the pancreas: Based on a clinical case.
ABSTRACT
The Autoimmune pancreatitis (AIP) is a type of chronic pancreatitis characterized histologically by a lymphoplasmocytic infiltration and periductal fibrosis. The clinical presentation may mimic a carcinoma of the head of the pancreas, especially in cases of cephalic disease, characterized by obstructive jaundice, abdominal pain and weight loss. This clinical entity may be associated with other autoimmune diseases and the diagnosis is based on the diagnostic criteria suggested by Mayo Clinic and the Japanese Society of the Pancreas, which associate the histological and radiological features to the increase in IgG4 and response to treatment with corticosteroids.
We present a case report of a patient who underwent a cephalic pancreatoduodenectomy, due to a preoperative diagnosis (clinical and radiological) of carcinoma of the head of the pancreas. The pathological study revealed that it was a lymphoplasmocytic pancreatitis and immunological study later confirmed the diagnosis of Auto Immune Pancreatitis.
This article aims to raise awareness for the differential diagnosis with carcinoma of the head of the pancreas, to avoid performing a surgery with relevant morbidity and mortality rates in a disease sensitive to treatment with corticosteroids.
KEY-WORDS: Autoimmune pancreatitis, carcinoma of the head of the pancreas, obstructive jaundice, lymphoplasmocytic infiltration, IgG4, cephalic pancreatoduodenectomy.
INTRODUÇÃO
A Pancreatite autoimune (PAI) é um tipo de pancreatite crónica também designada como pancreatite esclerosante linfoplasmocitária, pancreatite esclerosante, pancreatite inflamatória primária, pancreatite crónica com estreitamento irregular do ducto pancreático principal e pancreatocolangite esclerosante1.
A designação de PAI foi-lhe atribuída por Yoshida and al2 em 1995, com base na sua etiopatogenia.
A prevalência da doença está estimada em cerca de 4 a 11%3, sendo mais frequente no homem3 (82% dos doentes)4, na quinta5, sexta e sétima décadas da vida3.
Clinicamente a PAI pode mimetizar o carcinoma da cabeça do pâncreas6, principalmente se a doença se limitar ao segmento cefálico (80% dos doentes)3, sendo o quadro clínico dominado por icterícia obstrutiva, dor abdominal e perda ponderal.1,3 Cerca de 43 a 68% dos doentes com PAI apresentam diabetes mellitus.5,7,8
Até muito recentemente a maioria dos doentes com PAI eram submetidos a uma intervenção cirúrgica; já que o diagnóstico só muito raramente era efectuado antes da análise histopatológica da peça operatória. Actualmente, sabe-se que os corticóides podem induzir uma remissão completa da doença: clínica, radiológica e serológica; embora possam ocorrer recidivas após a interrupção da corticoterapia (10% dos casos)2,17,19,20.
Tendo tido oportunidade, recentemente, de tratar um caso clínico de PAI em que o diagnóstico não foi feito antes da intervenção, decidimos dá-lo a conhecer com o objectivo de recordar esta entidade nosológica pouco comum.
MATERIAL E MÉTODOS
Apresenta-se o caso clínico de uma doente do sexo feminino, de 59 anos de idade, com dor abdominal localizada ao epigastro com uma semana de evolução, associada a icterícia obstructiva e vómitos alimentares. Referia anorexia, astenia e emagrecimento (12 Kg no último ano, correspondendo a 17 % do peso). Apresentava, ao exame objectivo, um IMC de 20.
Como antecedentes pessoais relevantes refira-se a presença de diabetes mellitus desde há cerca de 10 anos e que se tornara insulinodependente no último mês. A restante história pessoal e familiar eram irrelevantes.
O abdómen era doloroso à palpação do epigastro, mas não existiam outras alterações significativas no exame objectivo. Analiticamente, apresentava hiperbilirrubinémia (9,5 mg/dl, com 5,8 mg/dl de bilirrubina directa), Aspartato aminotransferase (AST) de 458 U/L, Alanina aminotransferase (ALT) de 264 U/L, Gama-Glutamil transpeptidase (G-GT) de 383 U/L e Fosfatase Alcalina (FA) de 1615 U/L. A bilirrubinemia diminuiu durante o internamento (para 1,3 mg/dl com 0,8 mg/dl de bilirrubina directa), mantendo-se o restante quadro clínico e laboratorial.
A ecografia abdominal revelou dilatação das vias biliares intra e extra hepáticas, sem litíase vesicular ou da via biliar principal e a presença de um nódulo com 37 mm, de provável etiologia tumoral, na cabeça do pâncreas. A Tomografia computorizada (TC) toraco-abdominal confirmou a dilatação das vias biliares intra e extrahepáticas (12 mm), a existência duma cabeça do pâncreas globosa (4cm de maior eixo), isodensa e heterogénea, sem ectasia do Wirsung; e, ainda, uma trombose parcial da veia mesentérica superior. O quadro imagiológico era fortemente sugestivo de neoplasia da cabeça do pâncreas. (Figura 1)
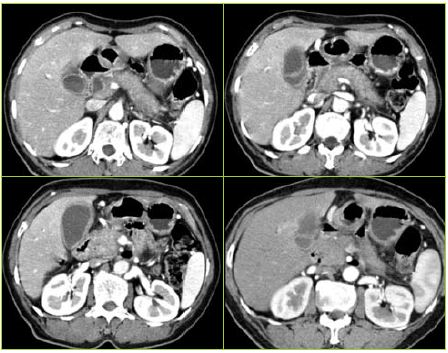
Figura 1 TC Abdominal: Efeito de realce heterogéneo da cabeça do pâncreas que revela dimensões aumentadas e determina dilatação da via biliar principal a montante, sugerindo malignidade. Alargamento de toda a glândula e presença de um halo hipodenso, não associados a dilatação do canal pancreático principal.
Os marcadores tumorais eram negativos. Uma endoscopia digestiva alta permitiu excluir a hipótese de ampuloma.
Tendo em conta o quadro clínico, laboratorial e radiológico, foi proposta para duodenopancreatectomia cefálica.
Durante a intervenção cirúrgica, tendo em conta a consistência pétrea de toda a glândula foram realizadas biópsias extemporâneas da cabeça e cauda pancreáticas que revelaram alterações compatíveis com pancreatite crónica (fibrose e infiltrado linfoplasmocitário) e ausência de células neoplásicas. Realizou-se, contudo, tendo em conta a marcada dilatação da via biliar principal e, também, à existência de trombose parcial da veia mesentérica superior, uma operação de Whipple clássica com anastomose pancreático-jejunal termino-lateral.
RESULTADOS
O pós-operatório decorreu sem complicações, com melhoria clínica e laboratorial. O diagnóstico histopatológico da peça operatória foi de pancreatite esclerosante linfoplasmocitária de provável etiologia autoimune. (Figura 2)
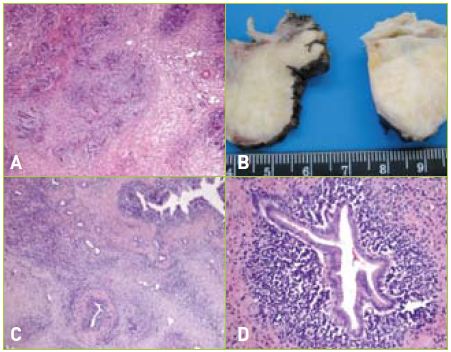
Figura 2 A. Biópsia extemporânea: evidência de fibrose e infiltrado inflamatório crónico. Ausência de células neoplásicas B. Peça operatória de duodenopancreatectomia cefálica que revela lesão superficialmente multinodular e de consistência pétrea C e D. Exame histopatológico: presença fibrose e infiltrado linfoplasmocitário compatível com PAI.
Foi então efectuado o estudo imunológico da doente que demonstrou elevação da IgG4 (1,59 g/dl) e da IgE (146 UI/ ml). Os auto-anticorpos anti-nucleares (ANA), anti-anidrase carbónica II (ACA II), anti-lactoferrina (ALF) e o factor reumatoide (FR) ALF eram negativos.
A doente encontra-se clinicamente bem 27 meses após a intervenção não existindo sinais clínicos, biológicos ou imagiológicos de recidiva.
DISCUSSÃO
A apresentação clínica, laboratorial e radiológica da PAI, principalmente quando sob a forma de doença segmentar cefálica (80% dos doentes3), pode mimetizar, como já ficou expresso, um quadro de adenocarcinoma da cabeça do pancreas3,6. Cerca de 2%3 a 5%22 das duodenopancreatectomias são realizadas em doentes com PAI, devido ao diagnóstico pré-operatório, não histológico, de neoplasia da cabeça do pâncreas.
A etiopatogenia da PAI, não se encontra ainda totalmente esclarecida, embora pareça ser condicionada por mecanismos autoimunes, tanto celulares (envolvendo linfócitos T CD4+), como humorais1.
Hamano et al9, em 2001 descreveram a elevação da IgG4 como um marcador sensível e especifico para PAI; os níveis de IgG4 estariam aliás associados à actividade da doença.
Kenji Hirano et al em 200510, num estudo que envolveu 116 pacientes do Hospital Universitário de Tóquio observaram valores elevados da IgG4 sérica em 94% dos doentes com PAI; que não encontraram noutras doenças pancreáticas malignas ou biliares.
Os níveis de auto-anticorpos ANA, ACA II, ALF e o FR encontram-se, também, muitas vezes elevados nesta entidade nosológica2,5,11,12,13.
Aliás o aparecimento frequente de manifestações extrapancreáticas (38%14) sugere a existência de antigénios alvo comuns ao pâncreas, glândulas salivares, vias biliares, túbulos renais, retroperitoneu e nódulos linfáticos mediastínicos.
A PAI encontra-se associada, em cerca de 20 a 40% dos doentes, a outras doenças autoimunes como o Sindroma de Sjogren, a cirrose biliar primária, a colangite esclerosante primária, as doenças inflamatórias intestinais e o lúpus eritematoso sistémico3,5,11,12,15,16. No entanto, a nossa doente não apresentava quaisquer destas situações clínicas.
Histológicamente esta entidade clínica é caracterizada pela presença de um infiltrado linfoplasmocitário, inflamação periductal, fibrose intersticial e periflebite. O pâncreas, bem como os outros órgãos envolvidos na PAI, apresentam infiltração tecidual com células positivas para IgG414.
A TC permite, habitualmente, pôr em evidência um aumento difuso ou focal da glândula, hipoatenuação do tecido adiposo peripancreático (resultante das alterações fibro-inflamatórias) e alterações irregulares do Wirsung; sem que exista a dilatação ductal típica do carcinoma pancreático3.
A colangioressonância magnética (CPRM) pode revelar alterações das vias biliares e ductos pancreáticos; no entanto a Colangiopancreatografia Retrógrada Endoscópica (CPRE) tem maior sensibilidade, podendo revelar estenose segmentar ou difusa do Wirsung, sem dilatação a montante2,17.
A ecoendoscopia demonstra, neste tipo de patologia, um pâncreas difusamente aumentado e hipoecogénico, ou uma massa solitária na cabeça do pâncreas. Este método de diagnóstico revelou-se superior à TC, CPRM e CPRE para a detecção de massas pancreáticas de pequenas dimensões. Quando associada a biópsia por agulha fina tem uma precisão de 85 a 96% no diagnóstico diferencial entre massas pancreáticas benignas e malignas18. Além disso a core biópsia guiada por ecoendoscopia revela, nestes doentes, infiltrados linfoplasmocitários positivos para IgG414. No nosso caso a doente realizou endoscopia digestiva alta com o intuito de excluir a presença de um ampuloma, uma vez que apresentou diminuição da hiperbilirrubinémia durante o internamento; no entanto a ecoendoscopia com biópsia não foi feita já que não se colocou a hipótese diagnóstica de PAI.
Tendo como objectivo minimizar o risco dum falso diagnóstico de PAI Suresh T. Chari et col, da Clínica Mayo, defenderam que este se deveria basear em três grupos de critérios diagnósticos: características histológicas típicas como a infiltração tecidular com IgG4, resposta da doença à corticoterapia e, ainda, outros critérios serológicos e radiológicos já incluídos, aliás, nos critérios da Sociedade Japonesa do Pâncreas, estabelecidos com o mesmo objectivo (Quadros 1 e 2).
Quadro 1 Critérios Diagnósticos clínicos para PAI do Japanese Research Committee of Intractable Diseases ofthe Pâncreas.

Quadro 2 Grupos diagnósticos para a PAI (Experiência da Clínica Mayo)

No caso clínico descrito a validação destes critérios só pôde ser feita no pós-operatório uma vez que o estudo histológico e imunológico somente foi realizado nessa altura.
Contudo, embora tardiamente, permitiu o diagnóstico de PAI na nossa doente já que esta apresentava uma hipertrofia da glândula mais acentuada na sua porção cefálica, associada a elevação da IgG4 sérica e a fibrose pancreática com infiltração linfoplasmocitária.
Os critérios diagnósticos da Sociedade Japonesa do Pâncreas e da Clínica de Mayo para a PAI, foram estabelecidos com o intuito de clarificar o diagnóstico desta entidade clínica.
Na PAI a icterícia é flutuante em cerca de um terço dos doentes e os marcadores tumorais nunca sofrem uma elevação tão marcada como no carcinoma da cabeça do pâncreas1; como, aliás, se verificou na nossa doente.
Hardacre et al16 referem ser tecnicamente mais difícil este tipo de cirurgia nos doentes com PAI, devido à reacção fibrótica peripancreática e que o número de complicações pós-operatórias é significativamente superior.
Toomey DP et al em 2007 propuseram um algoritmo diagnóstico para a PAI, na tentativa de evitar que doentes com este tipo de patologia fossem submetidos a uma cirurgia extensa como a DPC; mas que, simultaneamente, evitasse que se subestimasse uma possível lesão neoplásica (Figura 3).
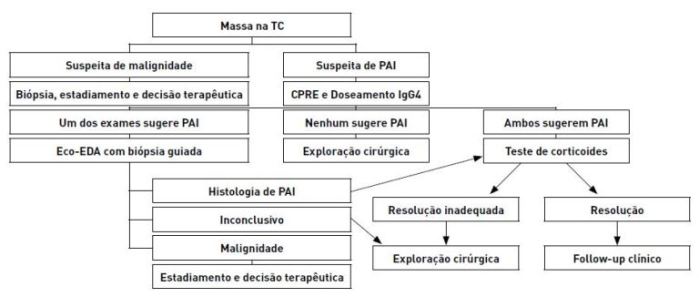
Figura 3 Algoritmo diagnóstico da Pancreatite Autoimune. Adaptado de Toomey DP et al3. PAI, Pancreatite Autoimune; TC, Tomografia computurizada; Eco-EDA, Ecoendoscopia; CPRE, colangiopancreatografia retrógrada endoscópica.
A TC da nossa doente não revelava irregularidades do Wirsung mas o aspecto imagiológico da massa heterogénea presente na cabeça do pâncreas era fortemente sugestivo de neoplasia. No entanto, caso nos tivéssemos orientado pelo algoritmo de Toomey DP et al a presença de uma lesão sugestiva de malignidade, condicionaria a realização duma biópsia (que não realizamos sistematicamente devido aos riscos de disseminação tumoral) cujo resultado provável seria uma histologia compatível com PAI, o que teria condicionado a realização da prova terapêutica com corticóides. Com efeito pensa-se, actualmente, que qualquer massa pancreática, diagnosticada como uma provável PAI, que não apresente resolução após duas a quatro semanas de corticoterapia, deverá ser explorada cirurgicamente; evitando atrasos no tratamento duma possível neoplasia21.
REFERÊNCIAS
1. Ketikoglou I, Moulakakis A. Autoimmune pancreatitis. Dig Liver Dis 2005;37:211-215. [ Links ]
2. Yoshida K, Toki F, Takeuchi T, et al. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995;40:1561-1568. [ Links ]
3. Toomey DP, Swan N, Torreggiani W, et al. Autoimmune pancreatitis. Br J Surg 2007;94:1067-1074. [ Links ]
4. Fernandez-del Castillo CF, Sahani DV, Lauwers GY. A 36-year-old man with recurrent epigastric pain and elevated amylase levels. N Engl J Med 2003;349:893-901. [ Links ]
5. Okazaki K, Chiba T. Autoimmune related pancreatitis. Gut 2002;51:1-4. [ Links ]
6. Khalili K, Deirdre JD, Chawla TP, et al. Renal cortical lesions in patient with autoimmune pancreatitis: a clue to differenciation from pancreatic malignancy. Eur J Radiol 2007; doi: 10.1016/j.ejrad.2007.07.020. [ Links ]
7. Ito T, Nakano IT, Koyanagi S, et al. Autoimmune pancreatitis as a new clinical entity: three cases of autoimmune pancreatitis with effective steroid therapy. Dig Dis Sci 1997;42:1458-1468. [ Links ]
8. Okazaki K, Uchida K, Chiba T. Recent concept of autoimmune-related pancreatitis. J Gastroenterol 2001;36:293-302. [ Links ]
9. Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344:732-738. [ Links ]
10. Hirano K, Kawabe T, Yamamoto N, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clinica Chimica Acta 2006;367:181-184. [ Links ]
11. Kloppel G, Luttges J, Sipos B, et al. Autoimmune pancreatitis: pathological findings. JOP 2005;6(suppl): 97-101. [ Links ]
12. Okazaki K, Uchida K, Ohana M, et al. Autoimmune related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000;118:573-581. [ Links ]
13. Kim K-P, Kim M-H, Kim JC, et al. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 2006;12:2487-2496. [ Links ]
14. Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol 2006;4:1010-1016. [ Links ]
15. Okazaki K, Kawa S, Kamisawa T, et al. Clinical diagnosis criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol 2006;41:626-631. [ Links ]
16. Hardacre JM, Iacobuzio-Donahue CA, Sohn TA, et al. Results of pancreaticoduodenectomy for lymphoplasmacytic sclerosing pancreatitis. Ann Surg 2003;237:853-858. [ Links ]
17. Horiuchi A, Kawa S, Hamano H, et al. ERCP features in 27 patients with autoimmune pancreatitis. Gastrointest Endosc 2002;55:494-499. [ Links ]
18. Farrel JJ, Garber J, Sahani D, et al. EUS findings in patients with autoimmune pancreatitis. Gastrointest Endosc 2004;60:927-936. [ Links ]
19. Kamisawa T, Egawa N, Nakajima H, et al. Clinical difficulties in the differentiation of autoimmune pancreatitis and pancreatic carcinoma. Am J Gastreoenterol 2003;98:2694-2699. [ Links ]
20. Nishino T, Toki F, Oyama H, et al. Long-term outcome of autoimmune pancreatitis after oral prednisolone therapy. Intern Med 2006;45:497-501. [ Links ]
21. Finkelberg D, Sahini D, Deshpande V, et al. Autoimmune pancreatitis. N Eng J Med 2006;355:2670-2676. [ Links ]
22. Van Gulik TM, Reeders JW, Bosma A, et al. Incidence and clinical findings of benign, inflammatory disease in patients ressected for presumed pancreatic head cancer. Gastrointest Endosc 1997;46:417-423. [ Links ]
*Autor para correspondência
Hospitais da Universidade de Coimbra
Serviço de Cirurgia III
Morada: Rua Dr. Pereira Caldas, nº205, 2º Esquerdo Frente – 4815 Vizela – Portugal
Telemóvel: +351 936 291 471
E-mail: juliana.c.oliveira@gmail.com
Recebido para publicação: 31/12/2009 e Aceite para publicação: 06/06/2010
