










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkJornal Português de Gastrenterologia
versão impressa ISSN 0872-8178
J Port Gastrenterol. vol.19 no.1 Lisboa jan. 2012
Hepcidina: A Molécula-Chave na Regulação do Metabolismo do Ferro
Graça Porto1,2, Susana Oliveira2, Jorge Pereira Pinto2
1Serviço de Hematologia Clínica, CHP-Hospital Santo António, Porto
2Basic and Clinical Research on Iron Biology, IBMC, Porto
*Autor para correspondência
RESUMO
A descoberta da hepcidina constitui uma das grandes revoluções do conhecimento na área da homeostasia do ferro. A demonstração de que este pequeno péptido de síntese maioritariamente hepática funciona como um controlador central do metabolismo do ferro e a subsequente elucidação dos seus mecanismos de acção e regulação, abriram novas portas na compreensão de um vasto leque de doenças do metabolismo do ferro e oferecem hoje novas perspectivas na prática clínica de diagnóstico e tratamento. Neste artigo são revistos alguns aspectos básicos da regulação e acção da hepcidina, e são também abordados novos conceitos que alargam o espectro de interesse à volta da hepcidina e da sua aplicação clínica.
PALAVRAS-CHAVE: Hepcidina, fígado, homeostasia do ferro, inflamação, anemia, sobrecarga de ferro
Hepcidin: the key-molecule in the regulation of iron metabolismo
ABSTRACT
The discovery of hepcidin constitutes one of the greatest revolutions in the present knowledge of iron homeostasis. The demonstration that this small peptide, synthesized mostly in the liver, functions as a central regulator of iron metabolism, and the subsequent elucidation of its mechanisms of action and regulation, all lead to a better understanding of a vast number of disorders of iron metabolismo and offer a new perspective in the diagnosis and treatment in the clinical practice. In this paper we review basic aspects of the regulation and action of hepcidin, and also address some novel concepts which enlarge the spectrum of interest around hepcidin and its clinical application.
KEY-WORDS: Hepcidin, liver, iron homeostasis, inflammation, anemia, iron overload
1. Introdução
O ferro é um elemento fundamental para um grande número de funções celulares, sendo a mais importante o transporte de oxigénio. Ao nível sistémico, o tecido que mais ferro consome é a medula óssea, que o utiliza no processo de síntese de hemoglobina pelos eritrócitos. A quantidade de ferro necessária para a eritropoiese é garantida através de um sistema eficiente de reciclagem no qual os macrófagos esplénicos fagocitam os eritrócitos senescentes, degradam a hemoglobina e exportam o ferro para a circulação para ser novamente utilizado. Se, por um lado, o ferro é um elemento vital para qualquer célula, ele pode ser também extremamente tóxico através da sua acção catalítica na produção de radicais livres de oxigénio. Por isso os seres vivos desenvolveram sistemas de regulação muito estritos do metabolismo do ferro, quer a nível de regulação do metabolismo intracelular quer ao nível sistémico, garantindo assim que este elemento não seja nem deficiente nem excessivo. As consequências do desequilíbrio do metabolismo do ferro são bem conhecidas clinicamente. De um lado do espectro clínico está a anemia por deficiência de ferro e do lado oposto as doenças de sobrecarga, em particular a Hemocromatose Hereditária (HH), na qual a acumulação excessiva de ferro pode levar eventualmente a lesão tecidular em vários órgãos, com manifestações como a cirrose hepática, diabetes, hipogonadismo, artrite ou cardiopatia.
2. A descoberta da Hepcidina
Pode dizer-se que a descoberta da hepcidina como o regulador central da homeostasia do ferro resultou da conjugação de vários estudos por grupos de investigação independentes que, num curto espaço de tempo isolaram, caracterizaram e demonstraram o seu efeito sistémico em modelos animais. A hepcidina foi originalmente isolada em dois estudos paralelos. Krause e colaboradores identificaram no ultrafiltrado de sangue humano um novo péptido de 25 aminoácidos rico em cisteínas, com propriedades antimicrobianas a que chamaram LEAP-1 (liver expressed antimicrobial peptide)1. Paralelamente, Park e colaboradores caracterizaram o mesmo péptido na urina humana, tendolhe chamado hepcidina por ser de síntese hepática (hep) e ter actividade antimicrobiana (cidin)2. A semelhança com o péptido descrito anteriormente no ultrafiltrado de sangue levou os autores a propor que o péptido tinha origem no fígado e atingia a urina através da circulação no plasma2. No mesmo ano Pigeon e colaboradores demonstraram que a produção do péptido era estimulada pela sobrecarga de ferro3 e, quase simultaneamente, Nicolas e colaboradores observaram que o silenciamento do gene da hepcidina em ratinhos transgénicos provocava um fenótipo de sobrecarga de ferro semelhante à hemocromatose humana4. No ano seguinte estes mesmos autores demonstraram que a sobre-expressão de hepcidina levava a uma deficiência severa de ferro com anemia, confirmando-se assim o papel fundamental da hepcidina como regulador dos níveis sistémicos de ferro5.
A hepcidina é essencialmente produzida nos hepatócitos, mas estudos mais recentes têm demonstrado que também é produzida por outras células, nomeadamente neutrófilos6, monócitos7-8, linfócitos9, adipócitos10, células beta do pâncreas11 e renais12. O papel da hepcidina de síntese extra-hepática não se encontra ainda completamente esclarecido, tal como a possibilidade da sua contribuição para o pool de hepcidina circulante, sendo provável que esta desempenhe um papel mais relevante na regulação de fluxos de ferro locais, funcionando num circuito autócrino e parácrino.
3. Regulação da produção de hepcidina
A hepcidina é sintetizada na forma de uma pré-próhormona, uma proteína de 84 aminoácidos biologicamente inactiva13. A maturação da hepcidina no hepatócito ocorre por clivagem da proteína sob a acção de uma convertase, a furina14-16, dando origem à sua forma activa, um péptido de 25 aminoácidos que circula no plasma e é excretado pela urina2. A expressão de hepcidina é regulada essencialmente por 4 tipos de sinais: 1) sinais de aumento da actividade eritropoiética; 2) sinais de resposta à concentração de ferro circulante; 3) sinais de resposta inflamatória e 4) sinais de stress retículo-endoplasmático (RE). Ao contrário dos dois primeiros tipos de sinal, cujos efeitos fisiológicos são muito evidentes, i.e., assegurar o aporte necessário de ferro para a eritropoiese ou proteger da deficiência ou toxicidade do ferro, não é ainda clara a importância biológica do aumento de hepcidina na inflamação ou em resposta ao stress RE. Uma compreensão mais aprofundada de todos estes mecanismos será essencial numa perspectiva de se prever quais as vantagens ou desvantagens de uma potencial intervenção terapêutica usando a hepcidina como alvo ou como agente. É de realçar que muitos dos mecanismos de regulação da hepcidina que se conhecem hoje foram esclarecidos com o estudo de modelos de doença, nomeadamente através da descoberta de mutações patogénicas em genes relevantes. Nos capítulos seguintes serão descritos mais detalhadamente esses mecanismos e tornar-se-á aparente que muitos dos sinais e vias de activação estão interligados, não havendo provavelmente processos de controlo completamente independentes ou que não sejam compensados por outros.
3.1. Regulação da hepcidina pela anemia, hipóxia e eritropoiese
A síntese de hepcidina é inibida em todas as situações de estímulo da actividade eritropoiética, tendo como efeito garantir a mobilização de ferro para a medula óssea.
Os mecanismos moleculares envolvidos nesta resposta são variados.
Na hipóxia, o principal sistema regulador envolve os chamados Hypoxia Inducible Factors (HIFs). Em condições de tensão normal de oxigénio, a subunidade α do HIF é hidroxilada e degradada através da via ubiquitina-proteossoma numa interacção com o gene supressor tumoral de von Hippel-Lindau (VHL). Pelo contrário, em situações de hipóxia ou de deficiência de ferro, a hidroxilação é inibida, resultando na estabilização da subunidade α do HIF e consequente activação de todos os genes que respondem especificamente a este factor de transcrição, como é o caso do gene da eritropoietina (EPO). Recentemente foi demonstrado que a hepcidina é um dos alvos do HIF-117, que se liga à região promotora do gene da hepcidina e diminui a sua transcrição.
A EPO é produzida pelo rim em condições de hipóxia ou deficiência de ferro, actuando, através da interacção com o seu receptor (EPOR), como o principal agente anti-apoptótico nos eritroblastos. A injecção de EPO é um potente inibidor da síntese de hepcidina in vivo18. O mecanismo, ou mecanismos, pelo qual a EPO actua como inibidor da hepcidina é ainda um assunto em investigação. Para além do efeito indirecto no aumento da eritropoiese, a EPO tem também um efeito directo de inibição da síntese de hepcidina pelos hepatócitos, através da regulação do factor de transcrição C/EBPα, mediada pelo EPOR19.
Um outro mediador da regulação negativa da hepcidina é o GDF15, um membro da família TGFβ de factores de crescimento, que se encontra muito elevado no soro de doentes com talassemia associado à expansão da massa eritrocitária21. Recentemente, foi também demonstrado que o GDF15 é estimulado em condições de deplecção celular de ferro, uma descoberta que apoia o facto de este factor estar também aumentado no soro de indivíduos deficientes em ferro ou após a administração de quelantes do ferro21.
Em condições de hipóxia existe também um aumento da concentração de radicais livres de oxigénio (ROS), o que também foi proposto como um mecanismo possível de repressão da expressão de hepcidina através da alteração das actividades do C/EBPα e STAT-322.
3.2. Regulação da hepcidina pelos níveis de ferro
Em condições de sobrecarga de ferro a hepcidina é induzida fisiologicamente para diminuir os níveis de ferro circulante e assim proteger da toxicidade do ferro3. A regulação da expressão da hepcidina em resposta ao ferro envolve outras moléculas, que funcionam como sensores de ferro e cujos mecanismos de acção explicam hoje a patogénese da maioria das doenças genéticas de sobrecarga de ferro, particularmente a HH.
A proteína HFE é o produto do gene responsável pela HH do adulto. Embora considerado um importante regulador da expressão da hepcidina, o seu mecanismo de acção ainda não está completamente esclarecido. O HFE interactua com os receptores da transferrina, TfR1 e TfR2, este último essencialmente expresso no fígado23. O complexo HFE-TfR2 (que se pensa ser formado em condições de elevada saturação da transferrina, por inibição competitiva da ligação HFETfR1) é um indutor da expressão da hepcidina através da via MAPK/ERK24. Não está ainda completamente esclarecido se o HFE é necessário como sensor da concentração de ferro ou se é apenas essencial para a produção basal de hepcidina25.
A proteína hemojuvelina (HJV) é o principal responsável pelas formas mais severas de Hemocromatose Juvenil e é um co-receptor de Bone Morphogenic Protein (BMP). As BMPs são membros da família TGFβ e induzem a transcrição do gene da hepcidina através da via das SMADs26. A HJV pode ser clivada pela furina (curiosamente, a mesma convertase responsável pela maturação da hepcidina) e libertada numa forma solúvel que, ao contrário da HJV de membrana, é capaz de inibir a produção de hepcidina, possivelmente por um mecanismo de competição com as BMPs.
Embora várias BMPs sejam capazes de induzir a expressão de hepcidina, a BMP6 é o mediador essencial para a resposta da hepcidina ao ferro no fígado27,28. Recentemente foi descrito que o HFE também interactua com a via de sinalização da BMP6 para regular a expressão de hepcidina, quer no modelo animal de deficiência de HFE29, quer em doentes com HH homozigóticos para a mutação C282Y do HFE30.
Uma nova molécula envolvida na via de activação HJV/BMP é a serina-protease TMPRSS6, também conhecida como matriptase-231. A deficiência de TMPRSS6 é a responsável pela anemia com deficiência de ferro resistente ao ferro, conhecida como IRIDA (iron-refractory iron deficiency anemia). A TMPRSS6 está envolvida na proteólise da HJV, sendo assim um inibidor da hepcidina32. Recentemente foi demonstrado que esta protease é também regulada por factores induzidos pela hipóxia33, evidenciando-se mais uma vez a inter-relação entre todos os mecanismos reguladores da hepcidina.
3.3. Regulação da hepcidina na inflamação e no stress RE
Possivelmente ligado à sua função como péptido antimicrobiano, a produção de hepcidina é induzida pela inflamação, sendo responsável pelo quadro de anemia da inflamação, também chamada anemia da doença crónica, num processo mediado pela IL-6 e envolvendo a via de sinalização JAK/STAT334,35. Outras citoquinas inflamatórias, tais como o TNF, poderão também estar envolvidas, mas o seu mecanismo ainda não é bem conhecido9.
O stress do RE é um processo bem conhecido nas células expostas a agentes tóxicos, alterações nutricionais ou outros agentes patológicos que interferem com o normal processamento das proteínas recém-sintetizadas no RE, induzindo um conjunto de respostas coordenadas conhecido como unfolded protein response (UPR). Esta resposta tem como objectivo manter o controlo de qualidade do RE e ajudar a célula a adaptar-se ou a recuperar de condições de stress. No entanto, esta resposta também tem sido apontada como um factor de progressão de doença em certos casos, como por exemplo na doença hepática crónica36. Dois artigos recentes mostraram que o stress RE em resposta a vários químicos é capaz de modular a expressão de hepcidina nos hepatócitos37,38.
3.4. Regulação da maturação da hepcidina
A maturação da hepcidina no hepatócito ocorre por clivagem da proteína sob a acção da furina, dando origem à sua forma activa, como um péptido circulante de 25 aminoácidos, e uma fracção pró-hepcidina. Os mecanismos exactos de regulação desta maturação são ainda muito mal conhecidos, sendo igualmente desconhecido o papel biológico da pro-hepcidina circulante. Muito recentemente foi demonstrado que o complexo HFE-TfR2, através da via de sinalização MAPK/ERK, regula a expressão de furina, podendo, desse modo, contribuir também para a regulação da maturação da hepcidina39.
4. A acção da hepcidina: o eixo hepcidina/ferroportina
O papel da hepcidina como regulador da homeostasia do ferro tem sido extensamente estudado, sendo claro que actua como regulador do influxo de ferro para o plasma a partir dos tecidos, nomeadamente dos enterócitos que absorvem o ferro da dieta, dos macrófagos que reciclam o ferro a partir dos eritrócitos senescentes, e dos hepatócitos que constituem o principal reservatório de ferro. Ao nível molecular, a hepcidina liga-se ao único exportador celular de ferro conhecido, a ferroportina, induzindo a sua internalização e degradação40,41. Mutações neste exportador estão na origem da chamada doença da ferroportina, uma forma de hemocromatose autossómica dominante e com características fenotípicas próprias que a distinguem de outras formas de HH42.
Embora a hepcidina tenha sido primeiro descrita como um péptido anti-microbiano, esta acção nunca foi bem caracterizada nos mamíferos. Pelo contrário, ao mediar a retenção de ferro nos macrófagos, a hepcidina pode ser responsável por favorecer o crescimento de microrganismos que utilizam este metal para o seu próprio crescimento43. Se a hepcidina circulante não parece ser eficiente como anti-microbiano, ainda está por esclarecer o seu papel quando é produzida pelas células do sistema imune. Recentemente foi descrito que também os linfócitos são capazes de produzir hepcidina que, ao inibir a ferroportina, induz a retenção do ferro, contribuindo assim para o controlo da sua própria proliferação9. A possibilidade de que a hepcidina sistémica possa contribuir para a activação e proliferação dos linfócitos à periferia é um assunto que está presentemente a ser investigado. Recentemente foi também proposto que a hepcidina poderá ter um outro papel na inflamação através da modulação da transcrição de citoquinas pró-inflamatórias44.
6. A importância da hepcidina na prática clínica
A descoberta da hepcidina e dos seus mecanismos reguladores veio explicar a patogénese de muitas doenças do metabolismo do ferro. No caso da HH, por exemplo, veio-se a esclarecer que todas as mutações descritas nos genes HFE, TfR2, HJV e HAMP têm um mecanismo patogénico comum que consiste na produção inapropriada, ou ausência de produção, de hepcidina em resposta ao excesso de ferro. Mas também em situações de sobrecarga de ferro adquirida, como nos casos da sobrecarga associada a doenças hepáticas crónicas como a hepatite C, ou a doença alcoólica, existe uma deficiência de produção de hepcidina45. Ao contrário, no fígado gordo nãoalcoólico (NASLD) existe um aumento da expressão da hepcidina46,47. Nestes casos, o aumento da hepcidina por inflamação crónica ou stress do RE favorece a depleção de ferro do sangue e a sua retenção nas células do sistema reticulo-endotelial, nomeadamente nas células de Kupffer, podendo estimular ainda mais a resposta imune e sendo assim um factor de agravamento da própria doença36. No caso da IRIDA, mutações no gene da TMPRSS6 são responsáveis pelo defeito de inibição da expressão da hepcidina em resposta ao ferro, sendo uma das características clínicas o aumento de hepcidina circulante48.
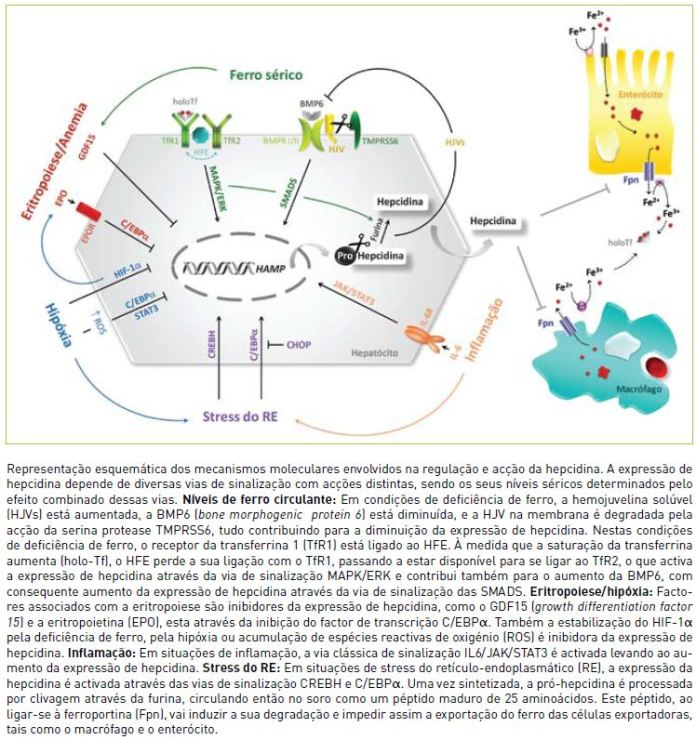
No contexto clínico, não há dúvida que o doseamento da hepcidina terá muito interesse no diagnóstico, classificação e seguimento dos doentes com as perturbações descritas do metabolismo do ferro, em particular no diagnóstico de formas raras de hemocromatose, na distinção entre uma hiperferritinémia de causa inflamatória ou por sobrecarga hepática de ferro e no diagnóstico clínico de IRIDA. Apesar do seu interesse clínico, não tem sido fácil implementar o doseamento da hepcidina na rotina laboratorial, sobretudo porque as metodologias que permitem medir concentrações das formas de hepcidina fisiologicamente relevantes são muito problemáticas e a sua disponibilidade para a comunidade médica que faz investigação clínica tem sido muito limitada. Os primeiros estudos de doseamento de hepcidina em modelos humanos foram realizados com um ensaio de doseamento de hepcidina urinária baseado na sua extracção da urina por um método de cromatografia de troca iónica com subsequente detecção por um anticorpo específico49. Este teste era muito laborioso e realizado apenas pelo próprio laboratório que produzia o anticorpo. Mais tarde foram introduzidas as técnicas de doseamento da hepcidina por espectroscopia de massa50,51, que têm uma boa reprodutibilidade e podem dosear a hepcidina não só na urina como também no soro, mas que dependem de equipamento muito dispendioso e que não se encontra facilmente disponível. Em termos de métodos imunoenzimáticos, foi descrito inicialmente um teste para detecção de pró-hepcidina, com um kit de ELISA comercialmente disponível52. No entanto, este teste nunca foi validado clinicamente já que os seus resultados não se relacionam com as concentrações esperadas de hepcidina-25 nem com outros parâmetros do metabolismo de ferro, não se sabendo ainda qual o significado fisiológico da pró-hepcidina53. Correlações significativas foram encontradas apenas na doença da ferroportina54, na doença renal terminal55 e na anemia da Paramiloidose Familiar (PAF)56. Recentemente foi descrito um método de ELISA competitivo que é capaz de detectar com precisão e reprodutibilidade alterações fisiológicas e patológicas nos níveis de hepcidina-25 no soro e urina57. No entanto a sua comercialização tem sido difícil, muito provavelmente pela dificuldade em garantir grandes stocks de anticorpo, dificuldade esta atribuída ao pequeno tamanho da molécula alvo (apenas 25 aminoácidos), à sua estrutura compacta e complexa, e à grande conservação genética entre espécies, o que torna difícil a indução de uma resposta imune nos modelos hospedeiros.
7. Conclusões
A descoberta da hepcidina revolucionou a área do metabolismo do ferro. Este péptido provou ser a hormona há muito tempo procurada, que funciona como o regulador central da homeostasia do ferro. A compreensão dos mecanismos da regulação da sua expressão colocaram o fígado no centro das atenções como o principal sensor das alterações sistémicas, e a compreensão do seu mecanismo de acção confirmou o papel das células exportadoras de ferro, tais como o epitélio intestinal e os macrófagos, como os principais efectores. Mas começa também a compreender-se que a hepcidina é produzida por muitos outros tipos celulares, podendo assim constituir uma proteína importante na regulação autócrina e parácrina do metabolismo celular do ferro.
O papel relevante da hepcidina na hemocromatose e noutras situações de alteração do metabolismo do ferro revelou que existe um conjunto muito heterogéneo de patologias que têm um mecanismo patogénico comum, a desregulação da hepcidina. Não há dúvida que o doseamento da hepcidina no soro será usado no futuro como uma ferramenta importante no diagnóstico, classificação e seguimento de doentes com patologias relacionadas com o ferro. No entanto, aguarda-se ainda pelo tempo em que possamos ter um teste robusto, comercialmente disponível e a um preço acessível, que torne o doseamento da hepcidina um teste de rotina na prática clínica. Finalmente, a hepcidina e os seus reguladores já são hoje vistos como potenciais alvos terapêuticos em patologias como a sobrecarga de ferro ou a anemia da doença crónica, embora para que tal possa ser uma realidade ainda haja necessidade de esclarecer muitos dos mecanismos moleculares da sua síntese, maturação, secreção e acção, e que permita uma compreensão plena da interacção entre todos os seus reguladores.
REFERÊNCIAS
1. Krause A, Neitz S, Mägert HJ, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 2000;480:147-150. [ Links ]
2. Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001; 276:7806-7810. [ Links ]
3. Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liverspecific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001; 276:7811-7819. [ Links ]
4. Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 2001; 98:8780-8785. [ Links ]
5. Nicolas G, Bennoun M, Porteu A, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA 2002; 99:4596-4601. [ Links ]
6. Peyssonnaux C, Zinkernagel AS, Datta V, et al. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006;107:3727-3732. [ Links ]
7. Liu XB, Nguyen NB, Marquess KD, et al. Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis 2005;35:47-56. [ Links ]
8. Sow FB, Florence WC, Satoskar AR, et al. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J Leukoc Biol 2007;82:934-945. [ Links ]
9. Pinto JP, Dias V, Zoller H, et al. Hepcidin Messenger RNA expression in human lymphocytes. Immunology 2010;130:217-230. [ Links ]
10. Bekri S, Gual P, Anty R, et al. Increased adipose tissue expression of hepcidin in severe obesity is independente from diabetes and NASH. Gastroenterology 2006;131:788-796. [ Links ]
11. Kulaksiz H, Fein E, Redecker P, et al. Pancreatic beta-cells express hepcidin, an iron-uptake regulatory peptide. J Endocrinol 2008;197:241-249. [ Links ]
12. Kulaksiz H, Theilig F, Bachmann S, et al. The ironregulatory peptide hormone hepcidin: expression and cellular localization in the mammalian kidney. J Endocrinol 2005;184:361-370. [ Links ]
13. Gagliardo B, Faye A, Jaouen M, et al. Production of biologically active forms of recombinant hepcidin, the iron-regulatory hormone. FEBS J 2008;275:3793-3803. [ Links ]
14. Valore EV, Ganz T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol Dis 2008;40:132-138. [ Links ]
15. Scamuffa N, Basak A, Lalou C, et al. Regulation of prohepcidin processing and activity by the subtilisin-like proprotein convertases Furin, PC5, PACE4 and PC7. Gut 2008; 57:1573-1582. [ Links ]
16. Kartikasari AE, Roelofs R, Schaeps RM, et al. Secretion of bioactive hepcidin-25 by liver cells correlates with its gene transcription and points towards synergism between iron and inflammation signaling pathways. Biochim Biophys Acta 2008;1784:2029-2037. [ Links ]
17. Peyssonnaux C, Zinkernagel AS, Schuepbach RA, et al. Regulation of iron homeostasis by the hypoxiainducible transcription factors (HIFs). J Clin Invest 2007;117:1926-1932. [ Links ]
18. Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002;110:1037-1044. [ Links ]
19. Pinto JP, Ribeiro S, Pontes H, et al. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood 2008;111:5727-5733. [ Links ]
20. Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 2007;13:1096-1101. [ Links ]
21. Lakhal S, Talbot NP, Crosby A, et al. Regulation of growth differentiation factor 15 expression by intracelular iron. Blood 2009;113:1555-1563. [ Links ]
22. Choi SO, Cho YS, Kim HL, et al. ROS mediate the hypoxic repression of the hepcidin gene by inhibiting C/EBPalpha and STAT-3. Biochem Biophys Res Commun 2007;356:312-317. [ Links ]
23. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996;13:399-408. [ Links ]
24. Ramey G, Deschemin JC, Vaulont S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 2009;94:765-772. [ Links ]
25. Lanzara C, Roetto A, Daraio F, et al. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood 2004;103:4317-4321. [ Links ]
26. Babitt JL, Huang FW, Xia Y, et al. Modulation of boné morphogenetic protein signaling in vivo regulates systemic iron balance.J Clin Invest 2007;117:1933-1939. [ Links ]
27. Meynard D, Kautz L, Darnaud V, et al. Lack of the boné morphogenetic protein BMP6 induces massive iron overload. Nat Genet 2009;41:478-481. [ Links ]
28. Kautz L, Meynard D, Besson-Fournier C, et al. BMP/ Smad signaling is not enhanced in Hfe-deficient mice despite increased Bmp6 expression. Blood 2009;17;114:2515-2520. [ Links ]
29. Corradini E, Garuti C, Montosi G, et al. Bone morphogenetic protein signaling is impaired in an HFE knockout mouse model of hemochromatosis.Gastroenterology 2009;137:1489-1497. [ Links ]
30. Ryan JD, Ryan E, Fabre A, et al. Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis. Hepatology 2010;52:1266-1273. [ Links ]
31. Ramsay AJ, Hooper JD, Folgueras AR, et al. Matriptase-2 (TMPRSS6): a proteolytic regulator of iron homeostasis. Haematologica 2009;94:840-849. [ Links ]
32. Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science 2008;320:1088-1092. [ Links ]
33. Lakhal S, Schoedel J, Townsend AR, et al. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxiainducible factors: new link between hypoxia signalling & iron homeostasis. J Biol Chem 2011; 286:4090-4097. [ Links ]
34. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006;108:3204-3209. [ Links ]
35. Verga Falzacappa MV, Vujic Spasic M, Kessler R, et al. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007;109:353-358. [ Links ]
36. Messner DJ, Kowdley KV. Biting the iron bullet: endoplasmic reticulum stress adds the pain of hepcidin to chronic liver disease. Hepatology 2010;51:705-707. [ Links ]
37. Oliveira SJ, Pinto JP, Picarote G, et al. ER stress-inducible factor CHOP affects the expression of hepcidin by modulating C/EBPalpha activity. PLoS One 2009;4:e6618. [ Links ]
38. Vecchi C, Montosi G, Zhang K, et al. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325:877-880. [ Links ]
39. Poli M, Luscieti S, Gandini V, et al. Transferrin receptor 2 and HFE regulate furin expression via mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/Erk) signaling. Implications for transferrin-dependent hepcidin regulation. Haematologica 2010;95:1832-1840. [ Links ]
40. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306:2090-2093. [ Links ]
41. De Domenico I, Ward DM, Langelier C, et al. The molecular mechanism of hepcidin-mediated ferroportin downregulation. Mol Biol Cell 2007;18:2569-2578. [ Links ]
42. Montosi G, Donovan A, Totaro A, et al. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001;108:619-623. [ Links ]
43. Paradkar PN, De Domenico I, Durchfort N, et al. Iron depletion limits intracellular bacterial growth in macrophages. Blood 2008;112: 866-874. [ Links ]
44. De Domenico I, Zhang TY, Koening CL, et al. Hepcidin mediates transcriptional changes that modulate acute cytokine-induced inflammatory responses in mice. J Clin Invest 2010;120:2395-2405. [ Links ]
45. Fujita N, Sugimoto R, Takeo M, et al. Hepcidin expression in the liver: relatively low level in patients with chronic hepatitis C. Mol Med 2007;13:97–104. [ Links ]
46. Harrison-Findik DD. Is the iron regulatory hormone hepcidin a risk factor for alcoholic liver disease? World J Gastroenterol 2009;15:1186–1193. [ Links ]
47. Mitsuyoshi H, Yasui K, Harano Y, et al. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol Res 2009;39:366–373. [ Links ]
48. Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA).Nat Genet 2008;40:569-571. [ Links ]
49. Nemeth E, Ganz T. Hepcidin and iron-loading anemias. Haematologica 2006;91:727-732. [ Links ]
50. Kemna EH, Tjalsma H, Podust VN, et al. Mass spectrometry-based hepcidin measurements in sérum and urine: analytical aspects and clinical implications. Clin Chem 2007;53:620-628. [ Links ]
51. Swinkels DW, Girelli D, Laarakkers C, et al. Advances in quantitative hepcidin measurements by time-of-flight mass spectrometry. PLoS One 2008;3:e2706. [ Links ]
52. Kulaksiz H, Gehrke SG, Janetzko A, et al. Pro-hepcidin: expression and cell specific localisation in the liver and its regulation in hereditary haemochromatosis, chronic renal insufficiency, and renal anaemia. Gut 2004;53:735-743. [ Links ]
53. Kemna EH, Tjalsma H, Willems HL, et al. Hepcidin: from discovery to differential diagnosis. Haematologica 2008;93:90-97. [ Links ]
54. Zoller H, McFarlane I, Theurl I, et al. Primary iron overload with inappropriate hepcidin expression in V162del ferroportin disease. Hepatology 2005;42:466-472. [ Links ]
55. Taes YE, Wuyts B, Boelaert JR, et al. Prohepcidin accumulates in renal insufficiency. Clin Chem Lab Med 2004;42:387-389. [ Links ]
56. Beirão I, Almeida S, Swinkels D, et al. Low sérum levels of prohepcidin, but not hepcidin-25, are related to anemia in familial amyloidosis TTR V30M. Blood Cells Mol Dis 2008;41:175-178. [ Links ]
57. Ganz T, Olbina G, Girelli D, et al. Immunoassay for human serum hepcidin. Blood 2008;112:4292-4297. [ Links ]
*Autor para correspondência
BCRIB, IBMC
Morada: Rua do Campo Alegre 823, 4150-180 Porto
E-mail: gporto@ibmc.up.pt
Recebido para publicação: 11/01/2011 e Aceite para publicação: 22/04/2011
