










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkJornal Português de Gastrenterologia
versão impressa ISSN 0872-8178
J Port Gastrenterol. v.17 n.3 Lisboa maio 2010
Zinc therapy of neurological Wilsons disease in a woman with two foetus with agenesis of the corpus callosum
António Murinello1, Teresa Tomé2, Augusto Goulão3, Álvaro Cohen2
1Hospital Curry Cabral – Internal Medicine Unit;
2Maternidade Alfredo da Costa – Centro de Diagnóstico Pré-Natal;
3RM – Caselas, Lisbon Portugal
Abstract
CLINICAL REPORT: A patient diagnosed Wilsons disease (WD) 22 years previously, successfully treated initially with zinc, developed neuropsychiatric disease after years of irregular therapy. Reassuming zinc therapy was successful. After a normal pregnancy, she had two therapeutic abortions for corpus callosum agenesis, and a missed abortion.
We review the genetics, physiopathology, clinics and imagiologic response to zinc therapy, the problems of pregnancy in WD, advising to maintain therapy. A hypothetic cause for fetus brain anomaly would be hypocupremia due to zinc therapy, confronting with two other possibilities, one related to Wilsons disease in itself, other due to a congenital syndrome of agenesis of the corpus callosum, impossible to diagnose by our available diagnostic methods.
Keywords: Wilsons disease, zinc, corpus callosum agenesis, hypocupremia.
Tratamento pelo zinco na fase neurológica da doença de Wilson numa mulher com dois fetos com agenesia do corpo caloso
Resumo
CASO CLÍNICO: Doente com doença de Wilson (DW) diagnosticada 22 anos antes, tratada inicialmente com sucesso com zinco, desenvolveu doença neuropsiquiátrica após anos de terapêutica irregular. A reintrodução do zinco foi eficaz. Após gravidez normal, teve duas interrupções terapêuticas por agenésias do corpo caloso fetal, e um caso de morte fetal.
Fez-se revisão da genética, fisiopatologia, resposta clínica e imagiológica à terapêutica com zinco, problemas da gravidez na DW, aconselhando-se manutenção terapêutica. Aventa-se hipótese da hipocuprémia induzida pelo zinco ser causal na anomalia fetal cerebral, em contraponto a outras duas possibilidades, uma relacionada com a própria DW, a outra a de ser resultante de uma das várias síndromes congénitas de agenésia do corpo caloso, não diagnosticáveis pelos meios de diagnóstico acessíveis aos autores.
Palavras-chave: Doença de Wilson, zinco, agenésia do corpo caloso, hipocuprémia.
INTRODUCTION
Progressive hepatolenticular degeneration of Wilsons disease (WD) is an inborn autosomal recessive disorder of copper metabolism, characterised by an inadequate excretion of absorbed dietary copper via bile, resulting in excessive accumulation of copper in the liver, central nervous system, and other organs, that can present clinically with hepatic, neurological or psychiatric disturbances, or a combination of these. Kayser-Fleischer rings result from copper deposition in Descemets membrane of the cornea. Mild or acute hemolysis may be a presenting feature. The most important determinant in WD symptomatology and its evolution appears to be the concentration of free copper in serum1.
The genetic defect in WD occurs on chromosome 13 at the level of ATP7B gene, which is 80 kilobases in length, consists of 21 exons and gives rise to a 165 kDa membrane bound protein (adenosine triphosphatase ATPase) with 1465 amino acids, with eight hydrophobic transmembrane sequences and six copper binding domains, a transduction domain, a cation channel and phosphorylation domain, and a nucleotide-binding domain2. The ATP7B protein is expressed primarily in liver but also in kidney, stomach, spleen, and other tissues3. As the typical daily intake of copper (0.6-1.6 mg a day) is more than the human body needs, a well functioning excretion must exist, in order to keep the organism from copper overindulgence leading to toxic effects. This is solved by the liver ATP7B protein, through incorporation of copper into caeruloplasmin for release to the circulation, but also necessary for excreting copper into bile. The exact mechanism of this process is unknown, but ATP7B protein appears to move to the canalicular membrane4,5. At January 2006, around 300 mutations have been described in the ATP7B gene, recorded in the Wilsons disease mutation database (http://www.medicalgenetics.med.ualberta.ca/wilson/index.php). Most patients are compound heterozygotes, partially explaining the variable spectrum of the disease6. Molecular genetic testing of the ATP7B gene is nowadays clinically available and is playing an increasingly important role in diagnosis, as copper studies are frequently equivocal, being sometimes essential for determining the genetic status of at risk sibs7.
The mutations cause a decrease or absence of the gene product or production of a protein with impaired function. This results in reduced copper transport within the hepatocyte and failed insertion of copper into caeruloplasmin. As a consequence, in most patients with WD serum caeruloplasmin and total copper serum concentrations are low, but there is an increased proportion of free low molecular weight copper species in serum, which deposits in excess in liver, brain, eyes, and other tissues. Absence of the protein in kidney impairs tubular reabsorption of filtered copper with resultant increased copper urinary excretion4,5.
According to the old paradigm copper accumulation would be noxius, causing symptoms due to organ dysfunction, but today it is accepted that symptoms are caused by copper poisoning due to increased free copper concentration in serum8. The accumulation of copper in the liver can be understood as a sign of detoxification of free copper in the liver by sequestration in a non-toxic metallothionein-bound form. If treatment aimed at lowering free copper concentrations in blood is started early, and if treatment is monitored correctly to ensure that free copper concentrations normalize, the symptoms may be completely reversible. If treatment is started later, severe liver disease and severe cerebral lesions may become irreversible.
Today it is generally accepted by several authors that treatment of copper poisoning with chelating agents is contraindicated, because it may lead to an increase in serum levels of free copper and iatrogenic clinical deterioration, sometimes irreversible8,9. Therapy with tetrathiomolybdate it is yet in a clinical experimental phase, being mostly useful for the treatment of severe neurological cases. Contrarywise to chelator therapy, zinc does not cause neurological deterioration. Some authors utilize zinc acetate 50 mg three times daily10 whereas others prescribe zinc sulphate 200 mg three times daily11. Zinc shall be taken 1 hour before or after meals. Zinc acts by blocking absorption of copper through induction of mucosal intestinal cell metallothionein12-15. Metallothionein has a greater affinity for copper than for zinc, and the metallothionein-copper complex is lost in stool after sloughing of intestinal epithelial cells15. Zinc also induces metallothionein synthesis at the liver. Brewer, et al16 demonstrated that no hepatic deterioration occurred in presymptomatic WD patients treated with zinc and followed for 3 to 9 years.
Copper is deposited throughout nearly all areas of gray and white matter of the central nervous system (CNS), but morphological changes are localized especially in the basal ganglia, deep cortical layers, cerebellum, brainstem and cerebral cortex17. MRI is more sensitive than CT to demonstrate the CNS lesions, particularly increased signal intensity on T2-weighted-images (T2WI) over the thalamus, basal ganglia and brainstem, especially the midbrain and pons. Regression of T2WI signal abnormalities on follow-up brain MRIs have been documented in several patients with WD on chelation treatment (D-penicillamine and trientine) and after liver transplantation18. Changes in MRI appropriate to clinical worsening or improvement on chelation treatment support copper redistribution either into or out of the CNS19. On serial MRI studies along 24 months of continuous treatment with zinc sulphate, Huang, et al20 demonstrated gradual resolution of the increased signal intensities in accordance with the clinical amelioration of the patients.
There are no contraindications to pregnancy in WD during maintenance therapy, with the exception of the presence of severe liver disease21, and anti-copper agents shall be continued during pregnancy. Otherwise women can suffer severe regression of their disease, often ending in death. Although some authors22,23 report favourable results of treatment of pregnant patients with WD, other authors refer to the possibility of occurrence of serious side effects of penicillamine24, favouring treatment with zinc. Also teratogeny has been documented in experimental animals and sometimes in humans with the chelating drugs penicillamine and trientine, the major factor in its production appears to be copper deficiency in the foetus25,26. Tetrathiomolybdate is not advised in pregnancy. According to several authors zinc therapy is considered essentially non toxic27-30. However Buamah, et al31 reports that low serum copper concentrations in pregnant woman during midgestation is a risk factor for foetal anencephaly, and Brewer, et al21 documented one case of foetal microencephaly and another of a newborn with minor surgically correctable heart defect in patients on treatment with zinc sulphate .
The authors present a case of a 38-year-old woman that, at the age of16, was diagnosed a hepatic form of WD32, responding favourably to zinc sulphate therapy during five years, but with neurological deterioration after irregular therapy for several years, and with clinical and imagiological amelioration after reassuming regular therapy with zinc sulphate. With therapy compliance the patient had one normal pregnancy and posteriorly two pregnancy terminations due to brain malformation (agenesis of the corpus callosum associated to ventriculomegaly). A possible correlation of these malformations with hypocupremia caused by zinc therapy is hypothesized. After that she had a missed abortion at 11 weeks of gestation, possibly related to Wilsons disease. Afterwards she decided to proceed to bilateral tubal ligation.
CLINICAL CASE
At 1986 a 16-year old portuguese girl was referred to us from the Hospital of Évora (Alentejo district) with a suspected diagnosis of chronic active hepatitis and hemolytic anemia. A diagnosis of hepatic form of WD was established based on clinical data, Kayser-Fleischer rings, abnormal liver function tests, low serum caeruleoplasmin, hypocupremia and hypercupruria (Table 1); the liver biopsy showed signs of chronic active hepatitis, liver fibrosis and nodular regeneration, and elevated concentration of copper on liver tissue: 536 μg/g of dry liver (N < 250 μg/g of dry liver). There were no neurologic symptoms and neurologic examination was normal. Central nervous system imaging was not performed. There were no other known cases of WD in her family. The patient was treated from then on with zinc sulphate (monohydrated salt) 200 mg three times daily. Five months later the patient was feeling much better and laboratory tests of copper biochemistry also showed favourable evolution (Table 1). One year after initiating the therapy the patient was asymptomatic, Kayser-Fleischer rings disappeared, liver function tests and also copper biochemistry were normal. Liver biopsy showed not only frank improvement of the inflammatory component, but also an almost normal concentration of copper on liver tissue (272 μg/g of dry liver). She was advised to maintain zinc therapy for life. Duing the next five years she did the therapy regularly, feeling well and being completely asymptomatic. Unfortunately the patient missed the appointments between 1991 and 1999, taking the medicine irregularly. In 1999 she left to German to work and failed to take the drugs for several consecutive days. From November to December 1999 she began to feel unwell, with asthenia, insomnia, dizziness, and strong headaches, with the sensation of a bigger and full head. She became progressively more depressed, crying frequently, having difficulties in personnel relationships, retiring frequently to dark rooms. She also noted progressive difficulty in making simple arithmetic calculus, and frank memory loss. She looks for us on January 2000. The MRI revealed lesions of the basal nucleus of the brain, compatible with the clinical diagnosis of neurological Wilson disease. High signal intensity lesions of the putamina on T2WI were present, suggesting edema and gliosis, as well as slight to moderate cerebral atrophy (Figs. 1 A, B).
Table 1. Serial evolution of copper metabolism along 22 years.
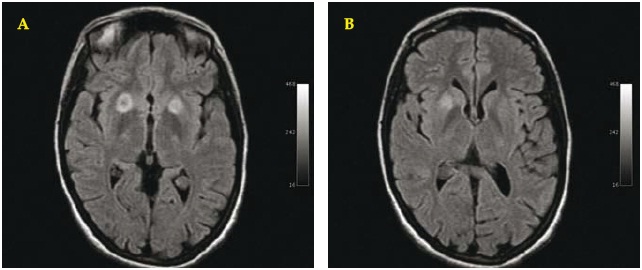
Fig.1 A/B. Brain MRI. January 2000. Axial T2-dark fluid WI: showing bilateral asymmetric hyperintense round target lesions of the putamina with greater expression on the right side (A).
LAB tests revealed: normal hemogram, blood coagulation and liver function tests, but some abnormalities of copper biochemistry (Table 1). We immediately initiated therapy with zinc sulphate 200 mg four times daily, that was maintained for one year, returning on that time to a dosage of 200 mg three times daily. Clinically the patient noticed progressive amelioration during the next three to four months, and asymptomatic from that point. She comes regularly to control until January 2001 (Table 1). Serial brain MRI were performed during the years (July 2000, December 2000, December 2003, July 2007), which revealed progressive decrease of the radiologic abnormalities, with just some residual aspects being found (Figs. 2 A, B).
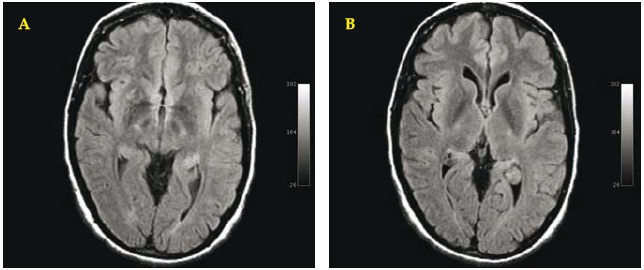
Fig. 2. A/B. Brain MRI. July 2007. Axial T2-dark fluid WI: revealing clear reduction of the hyperintensity of the lesions of the right putamen and essential normal aspect of the left putamen.
When the patient became pregnant in August 2002, the same zinc sulphate dosage was maintained. In October 2002 copper biochemistry was stabilized (Table 1). A boy was born in May 2003, without any perinatal problem; he is now four-years-old, and has a normal physical and intellectual development, and normal copper biochemistry. The patient continued the same treatment and by November 2003 copper biochemistry improved (Table 1). The patient didnt visit us, because she lives far from our city, but we know, by calling her that she maintained the same therapy and felt well. In September 2004 an interruption of pregnancy was performed on week 24 due to a prenatal diagnosis of agenesis of the corpus callosum associated with ventriculomegaly, confirmed by MRI at 22 weeks of gestation (Figs. 3 A, B). The amniocentesis realized just before the therapeutic abortion revealed a normal chromosomal study for the masculine sex. The necropsy of the foetus showed a complete agenesis of the corpus callosum and also a tetralogy of Fallot. No noxius drug was mentioned during pregnancy. Genealogic study of both father and mother families were negative for any suspected genetic disease related to the anomalies detected in the foetus. The patient was advised to take preconcepcional folic acid as she decided to get pregnant keeping the same dosage of zinc sulphate. However, at April 2007 and due to a prenatal ultrasound diagnosis of brain malformation, a complete agenesis of the corpus callosum associated with ventriculomegaly (Fig. 4), confirmed by MRI, another pregnancy interruption was done at 22 weeks of gestation. Again, no noxius drugs were used during pregnancy. Necropsy of the foetus revealed complete agenesis of the corpus callosum and also hydroureter and hydronephrosis of the right kidney. On June 2007 and May 2008 hemogram and blood coagulation as well as liver function tests were normal, but copper biochemistry was again slightly lifted up (adjusted copper concentration for caeruloplasmin and copper:caeruloplasmin ratio) (Table 1). For that reason we decided to advise the patient to take a zinc sulphate dosage of 200 mg four times daily and to ask the patient to follow a strict diet poor in copper (instead of her regular diet rich in copper). As by the 45th day copper biochemistry was again in normal limits, the dosage of 200 mg three times daily was advised once more. The patient, now 38-year-old, wishing to have a new pregnancy decided not to take a lower dosage of zinc therapy. At that time she found that was already pregnant of 11 weeks; in the first prenatal ultrasound the diagnosis of missed abortion was done. At that time the patient did a tubal ligation.
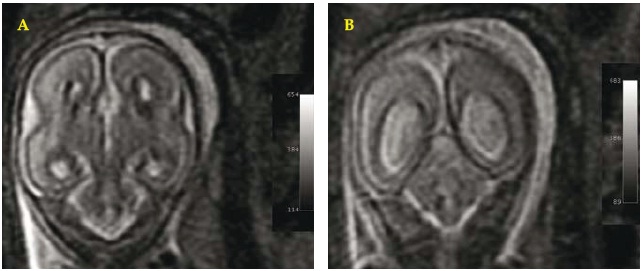
Fig. 3 A/B. Fetal brain MRI. September 2004 (22-23 weeks of gestation). Coronal WI: revealing typical aspects of agenesis of the corpus callosum: separation of the cerebral ventricles at the medial line (A,B) and colpocephaly of the ventricles (B).
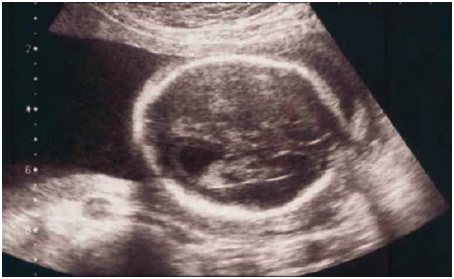
Fig.4. Prenatal ultrasonography. July 2008 (22 weeks of gestation): revealing fetal corpus callosum agenesis and ventriculomegaly.
DISCUSSION
Nonspecific imaging findings in the brain of WD patients include: general atrophy; abnormal gray matter of lentiform, caudate and thalamic nuclei; and abnormal infratentorial white matter35. Generalized atrophy suggests a more generalized susceptibility of the nervous system to copper toxicity. The specific neuroimaging findings of WD correspond to the neuropathologic spectrum of spongy to cystic degeneration of gray matter nuclei; central pontine myelinolysis; and softening, gliosis, and demyelination of nerve fibers36 Common abnormal MRI findings in WD patients, are increased signal intensity lesions on the T2 weighted images (T2WI) that are thought to reflect oedema, necrosis or cystic changes37,38. A characteristic, although infrequent finding, is the face of giant panda sign, due to T2 hyperintensity in the mesenchephalon that spares the red nucleus and pars reticulate of the substancia nigra39.
Wassener-van Hall, et al35 defines three types of abnormal gray matter signal intensities (SI) on MRI: type 1, high SI on T2W spin echo (SE) images, with any SI on T1WI; type 2, high SI on T1W inversion-recovery images, with any SI on T2W SE images; type 3, low SI on T2W SE images, with normal SI on T1W inversion-recovery images. In the same study, abnormal white matter was depicted with high SI on T2W SE and T2-weighted gradient-echo images and with mostly low SI on T1W inversion-recovery images. High SI in both gray and white matter on T2WI was the most common finding. MRI morphometry of the midbrain may be useful in the differential diagnosis of different neurodegenerative diseases. Through morphometric measures Semnic40 demonstrated prominent midbrain atrophy in patients with WD.
Abnormal high-signal-intensity lesions in the basal ganglia on T1WI can be seen in children with a clinical picture characterized mostly by liver disease and no severe neurologic symptoms. Possibly these abnormalities could be ascribed to biochemical changes associated with functional alterations that may precede the T2WI seen in more advanced forms of neurologic disease41. However the cause of isolated hyperintensity in T1WI remains elusive and Kozic42 suggested that this hyperintensity might be reversible during the course of chronic chelating therapy.
There are only a few reports regarding the fertility and outcome of pregnancy in WD. In the series of Sinha, et al43 recurrent abortions and stillbirths were common especially in women with untreated WD, but successful pregnancies and normal full-term delivery occurred in women receiving treatment and also in undiagnosed presymptomatic patients. Other authors44 found a high occurrence of successive unex plained abortions and advise the screening for WD in cases of unexplained recurrent abortions when family history demonstrated consanguinity or neurological, or psychiatric, and/or liver disease. The exact cause of these miscarriages is unclear, but it is believed that increased copper deposition in uterus prevents implantation of the fetus, due to free intrauterine copper derived from non-caeruloplasmin bound copper present in excess in these patients.
However, despite plausible toxicity of copper excess, copper is an essential trace element serving as an important catalytic cofactor in redox chemistry for a large number of proteins required for normal cell function. Copper deficiency gives rise to a multitude of symptoms, well illustrated by Menkes disease, an X-linked recessive disorder with a mutated ATP7A4.
There is increasing evidence that the origin of a significant number of developmental defects may be because of suboptimal nutrition during embryonic and foetal development45. Given the multiple roles that copper plays during foetal and embryonic development, the teratogenesis of copper deficiency likely involves an impairment of more than one pathway or process. The structural and biochemical defects associated with developmental copper deficiency must arise from several biochemical lesions, the significance of which being related to their temporal expression and severity46.
Studies in animals demonstrated that copper deficiency during embryonic and foetal development can result in numerous gross structural and biochemical abnormalities. Changes in free radical defence mechanisms, connective tissue metabolism, and energy production can all contribute to copper deficiency associated dysmorphogenesis. The biochemical alterations hypothetically underlying brain development abnormalities are: (1) reduction in the activity of the cuproenzyme, cytochrome-c oxidase; (2) excessive cellular oxidative damage through reductions in the activity of the oxidant defence enzyme-copper-zinc super oxide dismutase, resulting in peroxidation of lipids and oxidative damage to proteins and nucleic acids; (3) marked reduction of the brain cuproenzyme peptidylglycine monooxygenase, resulting in long-term behavioural and metabolic consequences in view of its impotence to hormone activation; (4) copper and low-molecular-weight copper complexes have angiogenic properties, being reasonable to speculate that altered angiogenesis may contribute to the brain dysmorphology associated with developmental copper deficiency; (6) markedly reduced protein-lysine 6-oxidase activity compromising vessel integrity, secondary to impaired collagen and elastin cross-linking and leading to haemorrhage and blood pooling in the embryonic fetal brain46.
The extent to which copper deficiency influences human prenatal development is not known. Buamah, et al31 demonstrated that low serum copper in non WD pregnant woman during midgestation was associated with an increased risk of anencephaly, and Morton, et al47 also demonstrated a correlation between low copper in drinking water and the occurrence of neural tube defects. Fosmire48 refer to the possibility of zinc overt toxicity symptoms with extremely high zinc intakes. At pharmacological intakes of zinc, copper deficiency is also induced and side effects can occur (anemia and leukopenia, abnormalities of immune response and of blood lipid profiles). Nevertheless zinc therapy is considered essential for the treatment of WD.
Consumption of high concentrations of zinc in the diet in experimental animals can induce a secondary copper deficiency as a consequence of decreased copper absorption and utilization49-52. Disease and drug-induced alterations in copper metabolism leading to copper deficiency may result in congenital malformations in experimental animals if maternal plasma copper level is low during the period of organogenesis. Studies in embryo culture systems in animals showed that copper-deficient embryo cultures in copper-deficient serum displayed numerous abnormalities including swollen hindbrain, blisters, blood pooling and distension of the large vessels. It is possible although not proved that copper deficiency at the conception has a causal effect on embryo malformations. It would be important to know if whether the effects of copper deficiency on the foetus or on the embryo are due directly to a deficiency of copper in foetal or embryonic cells, or whether they occur in part through indirect effects of copper deficiency on the metabolism of the mother53-58.
However at the level of clinical practice most authors consider that zinc therapy is essentially non toxic. Brewer, et al21 report 26 pregnancies of patients treated with zinc acetate resulting in 24 normal infants, 1 with a heart defect, and 1 microcephalic, concluding that zinc is the optimal choice for the pregnancies in women with WD. Notwithstanding the authors refer to the possibility that whatever the drugs utilized in mothers, the major risk of teratogenicity in WD pregnancies is overtreatment, reducing the copper level too low in the mother, and thereby affecting the foetus. However the risk appears to be greater with chelators due to the pulsatile nature of this therapy, in contrast with zinc that causes a gradual depletion of copper stores, avoiding blunt peaks of blood copper after mealtime. This levelling of blood copper levels, as opposed to peaks and through might be better physiologically for the fetus, and offer at least a theoretical advantage to zinc therapy. Due to the above considerations Brewer, et al21 reasons that physicians should perhaps aim for reasonable but not tight control of the mothers copper status during pregnancy. The authors advise to reduce the daily dosage of zinc acetate to half the dosage. We have no experience to advise the same attitude in treating these patients with zinc sulphate during pregnancy.
In this case the agenesis of the corpus callosum and the ventriculomegaly, occurred in association with congenital heart disease and with hydronephrosis. The corpus callosum starts its development at around 9 weeks, and at 20 weeks is normally fully developed. Its maldevelopment may be associated with other brain abnormalities like migrational disorders. The occurrence of the same clinical fenotype may be caused by an extrinsic mechanism, like a teratogenic mechanism or by a gene recessive autosomic or X-linked. The prenatal ultrasound and the necropsy didnt diagnose any other malformations, namely skeletal or facial. However the extension to which maternal dietary or drug-induced copper deficiency influences human pregnancy is not well understood.
In conclusion, we demonstrated that zinc therapy of neurological Wilsons disease is clinically and radiological effective, as was already proved by other authors, if the patients regularly comply with the advised therapeutic scheme. Pregnancy is associated with extraordinary metabolic demands on the mother and the developing foetus. Adequate maternal nutrition is essential for normal embryogenesis. In animals copper deficiency produces effects encompassing intrauterine growth retardation and teratogenic effect or embryonic death. We hypothesize that zinc-induced copper deficiency can be a mechanism for embryonic malformations. Eventually a lower dosage of zinc therapy, with strict control of the clinical evolution of the pregnant mother and of the laboratory tests related to copper metabolism, could be the most appropriate attitude in these patients.
ACKNOWLEDGMENTS
To Dr. J Pedro Alegria (Clinics of Diagnosis Dr. Fernando Teixeira) for the biochemical studies of the copper metabolism; to Dr. Eduardo Faustino (Invited Assistant of the Faculty of Pharmacy of the Lisbon University and Pharmacy of Alvide, in Cascais) for the manufacture of zinc sulphate tablets.
REFERÊNCIAS
1. Gaffney D, Fell GS, OReilly D. Wilson´s disease: acute and presymptomatic laboratory diagnosis and monitoring. J Clin Pathol 2000;53:807-812.
2. Frydman M, Bonne-Tamir B, Ferrer LA, et al. Assignement of the gene for Wilsons disease to chromosome 13. Linkage to esterase D locus. Proc Natl Acad Sci (USA) 1985;82:1819-1821.
3. Yamaguchi Y, Heiny ME, Shimizu N, et al. Expression of the Wilson`s gene is deficient in the Long-Evans Cinnamon rat. Biochem J 1994;301:1-4.
4. Waldenstrom E. Genetic and clinical studies in Wilsons disease. Acta Universitatis Upsala 2007. Digital Comprehensive Summaries of Upsala Dissertations from the Faculty of Medicine; pg 1-46.
5. Hoogenraad TU. From gene to disease: Wilsons disease: copper storage due to mutations in ATP7B. Ned Tijdschr Geneeskd 2003;147:1386-1387.
6. Thomas GR, Forbes JR, Roberts EA, et al. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet 1995;9:210-217.
7. Cox DW, Roberts E. Wilsons disease. Gene Reviews www.geneclinics.org/servlet/ Accessed in July 2010.
8. Hoogenraad TU. Paradigm shift in treatment of Wilsons disease: zinc therapy now treatment of choice. Brain and Development 2006;28:141-146.
9. Brewer GJ, Terry GA, Aisen AM, et al. Worsening of neurologic syndrome in patients with Wilsons disease with initial penicillamine therapy. Arch Neurol 1987; 44: 490-493.
10. Brewer GJ, Askari FK. Wilson´s disease: clinical management and therapy. J Hepatology 2005;42:S13-S21.
11. Hoogenraad TU, Van den Hammer CJ, van Hattum J. Effective treatment of Wilsons disease with oral zinc sulphate: two case reports. Brit Med J 1984;289:273-276.
12. Schouwink G. De hepato-cerebrale degeneratie (met een onderzoek naar de zinkstofwisseling). Amsterdam: University of Amsterdam, 1961. Thesis.
13. Sturniolo GC, Mestriner C, Irato P, et al. Zinc therapy increases duodenal concentration of metallothionein and iron in Wilsons disease patients. Am J Gastroenterol 1999;94:334-348.
14. Hill GM, Brewer GJ, Prasad AS, et al. Treatment of Wilsons disease with zinc: I. Oral zinc therapy regimens. Hepatology 1987;7:522-528.
15. Hoogenraad TU, Van Hattum J, Van de Hammer CJ. Management of Wilsons disease with zinc sulphate. Experience in a series of 27 patients. J Neurol Sci 1987;77:137-146.
16. Brewer GJ, Dick RD, Yuzbasiyan-Gurkan V, et al. Treatment of Wilsons disease with zinc XIII: Therapy with zinc in presymptomatic patients from the time of diagnosis. J Lab Clin Med 1994;123:849-858.
17. Starosta-Rubinstein S, Young AB, Kluin K, et al. Clinical assessment of 31 patients with Wilsons disease. Correlations with structural changes on magnetic resonance imaging. Arch Neurol 1987;44:365-377.
18. Nazer H, Brismar J, Al-Kawi MZ, et al. Magnetic resonance imaging of the brain in Wilsons disease. Neuroradiology 1993;35:130-133.
19. Heckman JM, Eastman RW, De Villiers JC, et al. Wilsons disease: neurological and magnetic resonance imaging improvement on zinc treatment. J Neurol Neurosurg Psychiatry 1994;57:1273-1274.
20. Huang CC, Chu NS. Wilsons disease: resolution of MRI lesions following long-term oral zinc therapy. Acta Neurol Scan 1996;93:215-218.
21. Brewer GJ, Johnson VD, Dick RD, et al. Treatment of Wilson´s disease with zinc. XVII: Treatment during pregnancy. Hepatology 2000;31:364-370.
22. Sternlieb I. Wilsons disease and pregnancy. Hepatology 2000;31:531-533.
23. Walshe JM. Pregnancy in Wilsons disease. Quart J Med 1977;46:73-83.
24. Marcellini M, di Ciommo V, Callea F, et al. Treatment of Wilsons disease with zinc sulphate from the time of diagnosis in pediatric patients: a single hospital, 10-year follow-up study. J Lab Clin Med 2005;145:139-143.
25. Cohen N, Keen CL, Lonnerdal B, et al. The effect of copper supplementation on the teratogenic effects of triethylenetetramine in rats. Drug-Nutr Interact 1983;2:203-10.
26. Keen CL, Lonnerdal B, Hurley LS. Drug-induced copper deficiency: a model for copper deficiency teratogenicity. Teratology 1983;28:155-156.
27. Brewer GJ, Yuzbasiyan-Gurkan V, Johnson V, et al. Treatment of Wilsons disease with zinc. XI. Interaction with other anti-copper agents. J Amer Coll Nutr 1993;12:26-30.
28. Hoogenraad TU. Wilsons disease. In: Major Problems in Neurology. London. WB Saunders 1996; vol 30.
29. Brewer GJ, Dick RD, Johnson V, et al. Treatment of Wilsons disease with zinc: long-term follow-up studies. J Lab Clin Med 1998;132:264-278.
30. Food and Drug Administration. Teratologic evaluation of FDA 71-49 (zinc sulphate). Food and Drug Research Laboratories, Inc. Prepared for Food and Drug Administration, United States Department of Commerce Publications PD-221 805, February 1973, and PB 267, June 1974.
31. Buamah PH, Russel M, Milford-Ward A, et al. Serum copper concentrations significantly less in abnormal pregnancies. Clin Chem 1984;30:1667-1667.
32. Murinello A, Ventura AM. Tratamento da doença de Wilson com sulfato de zinco. Revista de Gastrenterologia 19088; VI: 5-10 (in portuguese). [ Links ]
33. Twomey PJ, Viljoen A, House IM, et al. Adjusting copper concentrations for caeruloplasmin levels to routine clinical practice. J Clin Pathol 2006;59:867-869.
34. Twomey PJ, Viljoen A, House IM, et al. Copper: caeruloplasmin ratio. J Clin Pathol 2007;60:441-442.
35. Van Wassener-van Hall HN, Van den Heuvel AG, Algra A, et al. Wilson disease: findings at MR imaging and CT of the brain with clinical correlation. Radiology 1996;198:531-536.
36. Scheinberg HI, Sternlieb I. Neuropathology. In: Smith LH, ed. Wilsons Disease. 1rst ed. Philadelphia, Pa. Saunders; 1984:64-69.
37. Prayer L, Wimberger D, Kramer J, et al. Cranial MRI in Wilsons disease. Neuroradiology 1990;32:211-214.
38. Thomas KH, Aquilonius SM, Bergstrom K, et al. Magnetic resonance imaging of the brain in Wilsons disease. Neuroradiology 1993;35:134-141.
39. Giagheddu M, Tamburini G, Piga M, et al. Comparison of MR, EEG; EPs and ECHO-SPECT in Wilsons disease. Acta Neurol Scand 2001;103:71-81.
40. Semnic R, Svetel M, Dragosevic N, et al. Magnetic resonance imaging morphometry of the midbrain in patients with Wilsons disease. J Comput Assist Tomgr 2005;29:880-883.
41. Kim TJ, Kim IO, Kim WS, et al. MRI imaging of the brain in Wilsons disease of childhood: findings before and after treatment with clinical correlation. Am J Neuroradiol 2006;27:1371-1378.
42. Kozic D, Svetel M, Petrovic B, et al. MR imaging of the brain in patients with hepatic form of Wilsons disease. Eur J Neurol 2003;10:587-592.
43. Sinha S, Taly AB, Prashanth LK, et al. Successful pregnancies and abortions in symptomatic and asymptomatic Wilsons disease. J Neurol Sci 2004;217:37-40.
44. Schagen van Leeuwen JH, Christiaens GC, Hoogenraad TU. Recurrent abortion and the diagnosis of Wilsons disease. Obstetr Gynecol 1991;78:547-549.
45. Keen, CL Lonnerdal B, Hurley LS. Teratogenic effects of copper deficiency and excess. In, JR Sorenson, ed. Inflammatory Diseases and Copper. NJ, The Human Press, Clifton; 1982:109-121.
46. Keen CL, Uriu-Hare JY, Hawk SN, et al. Effect of copper deficiency on prenatal development and pregnancy outcome. Am J Clin Nutr 1998; 67 (suppl): 1003 S-1011 S.
47. Morton MS, Elwood PC, Albernety M. Trace elements in water and congenital malformations of the central nervous system in South Wales. Br J Prev Soc Med 1976;30:36-39.
48. Fosmire GJ. Zinc toxicity. Am J Clin Nutr 1990;51:225-227.
49. Reinstein NH, Lonnerdal B, Keen CL, et al. Zinc-copper interactions in the pregnant rat: fetal outcome and maternal and fetal zinc, copper and iron. J Nutr 1984;114:1266-1279.
50. Van Campen DR. Copper interference with the intestinal absorption of zinc by rats. J Nutr 1970;97:104-108.
51. Murty L, Klevay LM, HG Petering. Interrelationships of zinc-65 and copper nutriture in the rat. J Nutr 1974;104:1458-1465.
52. Mark-Savage P, Keen CL, Hurley LS. Reduction by copper supplementation of teratogenic effects of D-penicillamine. J Nutr 1983;113:501-510.
53. Turnlund JR. Copper. In, Shils ME, Olsen JA, Shike M, eds. Modern Nutrition in Health and Disease. 8th ed. Philadelphia, Lea & Febiger; 1994:231-241.
54. Uri-Hare JY, Stern YS, Keen CL. Influence of maternal dietary Zn intake on expression of diabetes-induced teratogenicity in rats. Diabetes 1989;38:1282-1290.
55. Zindenberg-Cheer S, Benak PA, Hurley LS, et al. Altered mineral metabolism: a mechanism underlying the fetal alcohol syndrome in rats. Drug Nutr Interact 1988;5:257-274.
56. Speich M, Murat A, Auget J-L, et al. Magnesium, total calcium, phosphorus, copper and Zn in plasma and erythrocytes of venous cord blood from infants of diabetic mothers: comparison with a reference group by logistic discriminant analysis. Clin Chem 1992;38:2002-2007.
57. Killholma P, Gronroos M, Erikkola R, et al. The role of calcium, copper, iron and zinc in preterm delivery and premature rupture of fetal membranes. Gynecol Obstet Invest 1984;17:194-201.
58. Artal R, Burgeson R, Fernandez FJ, et al. Fetal and maternal copper levels in patients at term with and without premature rupture of membranes. Obstetr Gynecol 1979;53:608-610.
Correspondência: António Murinello, Av.ª Eng.º Ant.º Azev.º Coutinho, Lt 8 r/c dto. 2750-644 Cascais. Portugal; E-mail: amurinello@yahoo.com; Tel.: +351 214 865 061; Tm.: +351 918 626 874.
Recebido para publicação: 23/12/2008 e Aceite para publicação: 12/05/2009.
