









Services on Demand
Journal
Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMedicina Interna
Print version ISSN 0872-671X
Medicina Interna vol.26 no.1 Lisboa Mar. 2019
https://doi.org/10.24950/rspmi/CC/153/1/2019
CASOS CLÍNICOS / CASE REPORTS
Uma Nova Variante no Gene Cacna1s Associada a Paralisia Periódica Hipocaliémica: Relato de um Caso
A Novel Variant in the Cacna1s Gene Associated with Hypokalemic Periodic Paralysis: A Case Report
Carla A Maia1
 https://orcid.org/0000-0002-6293-3100
https://orcid.org/0000-0002-6293-3100
Ana Hipólito-Reis1
https://orcid.org/0000-0003-0965-103X
Ana Oliveira Pinho1
https://orcid.org/0000-0001-5011-1954
Isabel Alonso2
https://orcid.org/0000-0001-8549-6903
J Vasco Barreto1
https://orcid.org/0000-0002-5391-5883
1Serviço de Medicina Interna, Unidade Local de Saúde de Matosinhos, Hospital Pedro Hispano, Matosinhos, Portugal
2Instituto de Investigação e Inovação em Saúde; CGPP e UnIGENe – Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugal .
RESUMO
A paralisia periódica hipocaliémica é uma doença rara, causada por variantes em canais de cálcio ou sódio dependentes de voltagem. Caracteriza-se por episódios frequentes de fraqueza muscular associados a hipocaliémia, frequentemente despoletados por situações de stress físico ou emocional. Relatamos o caso de um adulto do sexo feminino que se apresentou com um quadro de tetraparésia flácida e hipocaliémia grave, cuja caracterização genética permitiu identificar uma nova variante no gene CACNA1S. Após o diagnóstico iniciou terapêutica com acetazolamida, com redução da frequência e intensidade das crises. A paralisia periódica hipocaliémica constitui uma causa rara de fraqueza muscular súbita. Pode evoluir com fraqueza dos músculos respiratórios ou mesmo arritmias cardíacas, pelo que deve ser reconhecida de forma célere pelos clínicos.
Palavras-chave: Canais de Cálcio/genética; Paralisia Periódica Hipocaliémica; Mutação; Predisposição Genética para Doença
ABSTRACT
Hypokalemic periodic paralysis is a rare condition secondary to a genetic abnormality in calcium or sodium voltage dependent ion channels. It typically presents with muscle weakness and concomitant low serum concentration of potassium after exercise or emotional distress. We report the case of a woman who presented with flaccid tetraparesis and severe hypokalemia in which we have identified a novel probably disease-causing variant in the CACNA1S gene. After the diagnosis, the patient started acetazolamide treatment, resulting in a reduction in the intensity and number of episodes. Hypokalemic periodic paralysis is a rare cause of sudden muscle weakness. It may cause life-threatening respiratory involvement and cardiac arrhythmias, so all the clinicians should be aware of this condition in order to recognize it quickly.
Keywords: Calcium Channels/genetics; Genetic Predisposition to Disease; Hypokalemic Periodic Paralysis; Mutation
Introdução
A paralisia periódica hipocaliémica (PPH) é uma doença rara, com uma prevalência estimada de 1/100 000.1 Caracteriza-se por episódios recorrentes de fraqueza muscular associados a hipocaliémia grave e manifesta-se na maioria dos casos a partir da segunda década de vida.1,2 É causada por variantes patogénicas nos genes que codificam a subunidade de canais de cálcio (CACNA1S) ou sódio (SCN4A) dependentes de voltagem e transmite-se de forma autossómica dominante, com baixa penetrância nos indivíduos do sexo feminino, podendo também ocorrer de forma esporádica.2,3 Desconhece-se o mecanismo exato pelo qual variantes nestes genes resultam neste fenótipo; alguns estudos sugerem que resulta de alterações da expressão, localização subcelular e/ou cinética de canais de potássio não mutados, o que reduz as correntes de efluxo de potássio, resultando em hipocaliémia e despolarização patológica.4 Os episódios são frequentemente despoletados por infusões de glicose, refeições ricas em carbohidratos ou situações de stress físico/emocional, podendo ocorrer em intervalos de dias a anos.5 Cerca de 80% dos casos de PPH são causados por variantes no gene CACNA1S, e os restantes 20% por variantes no gene SCN4A.6 O gene CACNA1S é expresso nas células do músculo-esquelético humano e é responsável pela coordenação excitação/contração; o influxo de cálcio através deste canal provoca a libertação local de uma grande quantidade de cálcio através das reservas de cálcio intracelular, o que resulta na contração muscular.7
O tratamento profilático da PPH pode ser feito com suplementos de potássio, diuréticos poupadores de potássio ou inibidores da anidrase carbónica, como a acetazolamida.8 Desconhece-se o mecanismo pelo qual a acetazolamida é eficaz nesta patologia,9 mas parece ser mais eficaz nos casos de doentes com variantes causadoras de doença no gene CACNA1S.9
Apresentamos um caso de paralisia periódica hipocaliémica associado a uma variante no gene CACNA1S que não tinha sido previamente descrita na literatura.
Caso Clínico:
Um paciente do sexo feminino de 39 anos de idade recorreu ao Serviço de Urgência (SU) por fraqueza muscular de predomínio nos membros inferiores, de início súbito. Tinha sido submetida a cirurgia de libertação do túnel cárpico no mesmo dia. Referiu dois episódios prévios semelhantes: o primeiro aos 18 anos de idade, após exercício físico intenso, e o segundo aos 25 anos, sem fator despoletante evidente. Em ambas as ocasiões foi documentada hipocaliémia. Sem outros antecedentes médicos ou história familiar de relevo: a mãe, irmã e os dois filhos jovens nunca tiveram sintomas ou achados idênticos. O pai falecera aos 68 anos por complicações de traumatismo vertebro-medular, desconhecendo-se episódios idênticos. O estudo analítico no SU revelou hipocaliémia grave de 1,2 mEq/L (normal entre 3,5 e 5,5 mEq/L) e o eletrocardiograma mostrava prolongamento do intervalo QRS e aplanamento das ondas T. Sem alterações do equilíbrio ácido-base ou da função tiroideia. Ao exame físico destacava-se tetraparésia flácida de predomínio proximal. Iniciou de imediato reposição de potássio, com normalização da caliemia nas primeiras 24 horas e resolução progressiva dos sintomas. Após resolução da crise, manteve valores de potássio estáveis, entre 4,1-4,6 mEq/L. Não foi possível valorizar o gradiente transtubular de potássio, por estar há várias horas sob reposição de eletrólitos quando observada por nós. Foi orientada para a consulta externa de Medicina Interna.
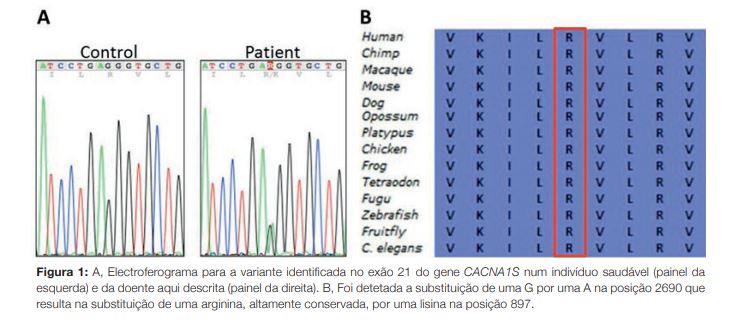
A realização de teste genético para pesquisa de variantes associadas a paralisia periódica hipocaliémica permitiu identificar a variante c.2690G > A;p. (Arg897Lys) em heterozigotia, ou seja, a substituição de uma guanina por uma adenina no codão 2690, que resulta na substituição de uma arginina, altamente conservada, por uma lisina na posição 897, localizada no segmento sensor de voltagem do canal (Fig 1). A análise bioinformática, utilizando software (SIFT, MutationTaster e UMD Predictor), indica que será patogénica. À data da detecção desta mutação (2014) a mesma não era conhecida. Mais tarde, em 2017, um laboratório com sede nos Estados Unidos da América registou a detecção da mesma mutação num indivíduo que também teria fenótipo de paralisia periódica hipocalémica.10 O significado clínico desta variante é ainda apoiado pelo facto de estar descrita outra variante causadora de doença no mesmo codão (codificando outro aminoácido), cujo fenótipo associado é de paralisia periódica hipocaliémica.11
A doente foi aconselhada em relação à evicção de fatores desencadeantes. Nos seis meses após o diagnóstico sofreu mais duas crises sintomáticas, tendo iniciado terapêutica com acetazolamida 250 mg uma vez por dia, que manteve durante 11 meses. Durante esse período sofreu um episódio clinicamente compatível com paralisia hipocaliémica que reverteu espontaneamente, sem confirmação laboratorial de hipocaliémia. Suspendeu posteriormente o fármaco (para planear uma gravidez), sem que se verificasse recorrência de crises. A ausência de resposta clinicamente evidente é compatível com relatos da literatura que referem taxas de resposta à acetazolamida da ordem dos 60% nos doentes portadores de mutações no gene CACNA1S,9 pelo que até ao momento se mantém sem terapêutica dirigida.
Realizado teste genético à mãe da doente, que não é portadora da variante. Os dois filhos e a irmã, assintomáticos até à data, não foram estudados.
Conclusão
A PPH constitui uma causa rara e pouco conhecida de fraqueza muscular súbita.12
Geralmente envolve apenas os músculos dos membros e as cinturas, mas existem casos descritos de morte por arritmias cardíacas e falência respiratória, pelo que deve ser reconhecida de forma célere pelos clínicos.12 A identificação de variantes genéticas associadas a entidades clínicas pode ser determinante para o desenvolvimento de terapêuticas dirigidas a alvos específicos.
Referencias
1. Lapie P, Lory P, Fontaine B. Hypokalemic periodic paralysis: An autosomal dominant muscle disorder caused by mutations in a voltage-gated calcium channel. Neuromuscul Disord. 1997;7:234–40.
2. Dogan NO, Avcu N, Yaka E, Isikkent A, Durmus U. Weakness in the Emergency Department: Hypokalemic Periodic Paralysis Induced By Strenuous Physical Activity. Turkish J Emerg Med . 2015;15:93–5.
3. Wang X, Ren B, Yong Z, Xu H, Fu Q, Yao H. Mutation analysis of CACNA1S and SCN4A in patients with hypokalemic periodic paralysis. Mol Med Rep. 2015;6267–74. http://dx.doi.org/10.3892/mmr.2015.4201
4. Kim JB, Kim JB, Kang SY, Yi JW, Kim SM. The large-conductance calcium-activated potassium channel holds the key to the conundrum of familial hypokalemic periodic paralysis. Korean J Pediatr. 2014;57:446–51. http://dx.doi.org/10.3345/kjp.2014.57
5. Kantola IM, Tarssanen LT. Familial hypokalaemic periodic paralysis in Finland. J Neurol Neurosurg Psychiatry. 1992;55:322–4.
6. Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. 2009;72:1544–7. http://dx.doi.org/10.1212/01.wnl.0000342387.65477.
7. Wang Q, Liu M, Xu C, Tang Z, Liao Y, Du R, et al. Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a Chinese family. J Mol Med. 2005;83:203–8.
8. Matthews E, Portaro S, Ke Q, Sud R, Haworth A, Davis MB, et al. Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype. Neurology. 2011;77:1960–4. http://dx.doi.org/10.1212/WNL.0b013e31823a0cb6.
9. Matthews E, Hanna MG. Muscle channelopathies: does the predicted channel gating pore offer new treatment insights for hypokalaemic periodic paralysis? J Physiol. 2010;588:1879–86. http://dx.doi.org/10.1113/jphysiol.2009.186627.
10. Ke Q, He F, Lu L, Yu P, Jiang Y, Weng C, et al. The R900S mutation in CACNA1S associated with hypokalemic periodic paralysis. Neuromuscul Disord. 2015; 25):955-8. http://dx.doi.org/10.1016/j.nmd.2015.09.006. [ Links ]
11. Stedwell RAYE, Allen KM, Binder LS. Hypokalemic Paralyses. Am J Emerg Med. 1992;10:143–8.
Correspondência:Carla A Maia carlanif@gmail.com
Serviço de Medicina Interna, Unidade Local de Saúde de Matosinhos, Hospital Pedro Hispano, Matosinhos, Portugal
Rua Dr. Eduardo Torres, 4464-513 Senhora da Hora
Conflitos de Interesse: Os autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
Fontes de Financiamento: Não existiram fontes externas de financiamento para a realização deste artigo.
Direito à Privacidade e Consentimento Informado: Os autores declaram que nenhum dado que permita a identificação do doente aparece neste artigo.
Proteção de Seres Humanos e Animais: Os autores declaram que não foram realizadas experiências em seres humanos ou animais.
Proveniência e revisão por pares: Não comissionado; revisão externa por pares.
Conflicts of interest: The authors have no conflicts of interest to declare.
Financing Support: This work has not received any contribution, grant or scholarship.
Confidentiality of data: The authors declare that they have followed the protocols of their work center on the publication of data from patients.
Protection of human and animal subjects: The authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Provenance and peer review. Not commissioned; externally peer reviewed
Recebido: 20/08/2018
Aceite: 31/10/2018

{kind=link}