










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMedicina Interna
versão impressa ISSN 0872-671X
Medicina Interna vol.25 no.2 Lisboa jun. 2018
https://doi.org/10.24950/rspmi/CC/254/2/2018
CASOS CLÍNICOS / CASE REPORTS
Envolvimento Intestinal Difuso por Macroglobulinemia de Waldenström: Caso Clinico e Revisão da Literatura
Diffuse Intestinal Involvement by Waldenström Macroglobulinemia: Clinical Case and Literature Review
Nídia Pereira1, Luís Miguel Afonso1, José Soares2, Mrinalini Honavar3, Maria João Santos4, J. Vasco Barreto1
1Serviço de Medicina Interna do Hospital Pedro Hispano, Matosinhos, Portugal
2Serviço de Gastrenterologia do Hospital Pedro Hispano, Matosinhos, Portugal
3Serviço de Anatomia Patológica do Hospital Pedro Hispano, Matosinhos, Portugal
4Unidade de Hematologia Clínica, Hospital Pedro Hispano, Matosinhos, Portugal
RESUMO
A marcroglobulinemia de Waldenström (MW) é uma doença linfoproliferativa pouco comum, sendo raros os casos de atingimento intestinal. Neste artigo descrevemos o caso de uma doente com atingimento intestinal difuso por marcroglobulinemia de Waldenström, cujas manifestações iniciais foram diarreia persistente com síndrome de má absorção, perda ponderal significativa e sintomas dispépticos. Os estudos endoscópicos revelaram atingimento de toda a mucosa do intestino delgado por depósitos de Imunoglobulina M e cadeias leves kappa. Posteriormente, a doente foi avaliada em consulta de Hemato-oncologia, apresentando baixo risco de mortalidade a 5 anos, pelo que não foram sugeridas propostas terapêuticas específicas. A doente permaneceu clinicamente estável até Dezembro de 2015, altura em que desenvolveu quadro de choque séptico com disfunção múltipla de órgãos de evolução rapidamente progressiva, acabando por falecer poucas horas após admissão no serviço de urgência. Os autores descrevem o caso desta doente e elaboram uma revisão da literatura sobre esta patologia. Pretende-se chamar a atenção para a necessidade de pensar na marcroglobulinemia de Waldenström como hipótese diagnóstica a considerar perante um doente com síndrome de má absorção.
Palavras-chave: Enteropatias; Imunoglobulina M; Macroglobulinemia de Waldenström; Síndromes de Malabsorção
ABSTRACT
Gastrointestinal involvement in Waldenström macroglobulinemia is an extremely rare lymphoproliferative disorder. We describe a Waldenström macroglobulinemia patient presenting with diarrhea, dyspepsia, weight loss and malabsorption. Endoscopic studies revealed diffuse deposition of immunoglobulin M and kappa light chains in the mucosa of the small intestine. Later, on evaluation in the Haemato-Oncology clinic, the risk of death at 5 years was considered low, and thus no further therapeutic measures were proposed. The patient remained stable for three years, when she developed septic shock with rapidly progressive multiple organ failure and died a few hours after admission to hospital. The authors describe the case and elaborate a review of the literature on this pathology. We draw attention to this rare manifestation of an uncommon lymphoproliferative disorder to be considered in the differential diagnosis in a patient with a malabsorption syndrome.
Keywords:Immunoglobulin M; Intestinal Diseases; Malabsorption Syndromes; Waldenström Macroglobulinemia.
Introdução
A macroglobulinemia de Waldenström (MW) é uma entidade rara que se define como uma doença linfoproliferativa monoclonal de células B,1 caracterizada pela presença de uma paraproteína IgM monoclonal no soro associada à identificação de populações celulares com características de linfoma linfoplasmocítico na medula óssea.1,2
O atingimento intestinal por MW é muito raramente descrito na literatura, tendo sido reportados cerca de 25 casos ao longo das últimas quatro décadas.1,3,4
Caso Clínico
Apresentamos o caso de uma doente do sexo feminino, de 48 anos, caucasiana, com diagnóstico de anemia hemolítica auto-imune por anticorpos frios em 2010. Nessa altura, identificação transitória de população monoclonal de linfócitos B na imunofenotipagem de sangue periférico e na medula óssea, compatível com linfoma linfoplasmocítico. Identificada também gamapatia monoclonal IgM/k sérica (pico M 0,89 g). Presença de hepatoesplenomegalia ligeira (baço 13 x 6,6 cm), sem evidência de adenomegalias. Sem sintomas relativos ao sistema gastrointestinal nessa data. Assumido o diagnóstico de linfoma não-Hodgkin de baixo grau, tendo sido realizada terapêutica com rituximab e corticóide com boa resposta, porém sem possibilidade de suspensão do segundo fármaco por recidiva da anemia. Por esse motivo a doente foi submetida a esplenectomia em Outubro de 2010, com resolução da anemia. Repetiu biópsia de medula óssea cujo resultado foi normal, excluindo alterações sugestivas de doença linfoproliferativa e indicando remissão após tratamento.
A doente foi posteriormente encaminhada para a Consulta de Medicina Interna, em Março de 2012, por um quadro de diarreia com 5 meses de evolução, associado a perda ponderal significativa (13 kg - 20,6% do peso corporal) e sintomas dispépticos. As dejecções tinham frequência variável (entre 1 e 5 dejecções diárias), por vezes com presença de alimentos não digeridos, sem sangue. Ao exame objectivo não foram evidentes alterações, nomeadamente adeno ou organomegalias.
Do estudo complementar efectuado salienta-se: elevação da velocidade de sedimentação (49 mm/1ª hora), ß2 microglobulina (4,53 mg/L, sendo o normal 0,97-2,64 mg/L) e dos níveis de IgM (1776 mg/dL); diminuição dos níveis de IgG (167 mg/dL) e de IgA (60 mg/dL). Evidente consumo de complemento (C3 71 mg/dL e C4 <2,9 mg/dL). Viscosidade sérica elevada (2,45 centipoise). Doseamento de alfa-1-antitripsina fecal elevada, com determinações de gordura fecal e elastase pancreática normais, favorecendo o diagnóstico de enteropatia perdedora de proteínas. Estudo imunológico a revelar apenas ANA positivos, com título de 1:320, padrão fino granular. Excluída doença celíaca. Mantoux a 2 unidades: 0 mm. Restante estudo analítico sem alterações, nomeadamente sem citopenias. Realizada tomografia computorizada (TC) abdominal que documentou hepatomegalia homogénea ligeira e espessamento difuso da parede das ansas do delgado. Colonoscopia sem alterações. Endoscopia digestiva alta a revelar mucosa duodenal nacarada, edemaciada, com perda do padrão vilositário (Fig 1). Cápsula endoscópica evidenciando transformação abrupta da mucosa após o piloro, com presença de placas coalescentes brancas e edema, associados a distorção das vilosidades, padrão que envolve toda a extensão do delgado (Fig 2).


O exame histológico de biópsia duodenal revelou atrofia vilositária parcial e expansão das extremidades das vilosidades por abundantes depósitos de material hialino positivo para a coloração PAS (mas negativo para vermelho de Congo) com expressão imunohistoquímica para IgM e cadeias leves kappa; sem evidência de linfoma (Fig 3). Foi observada linfangiectasia na lâmina própria. Estes achados são compatíveis com envolvimento difuso do intestino delgado por MW. Estudo microbiológico (incluindo parasitológico) foi negativo.
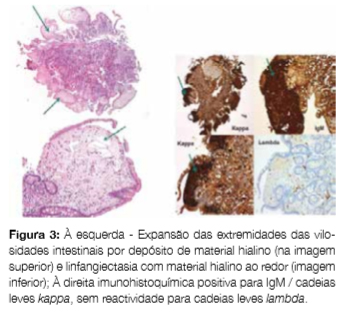
Foi também realizada biópsia de medula óssea que revelou infiltração medular por linfoma B periférico tipo linfoplasmocítico, sem rearranjo de imunoglobulinas.
A doente foi avaliada em consulta de Hemato-Oncologia de outro hospital, pontuando 1 no International Prognostic Scoring System for Waldenstöms Macroglobulinemia (ISSWM), correspondendo a baixo risco de mortalidade, sendo a taxa de sobrevivência a cinco anos estimada em 87%. Foi decidida vigilância clínica, sem propostas terapêuticas específicas (nomeadamente quimioterapia), consideradas nesta fase desproporcionalmente agressivas atendendo ao estado clínico da doente. Iniciou dieta adaptada, com alto teor de fibras solúveis e com suplementação proteica e calórica.
Permaneceu estável, mantendo apenas cuidados dietéticos. Apresentou clínica periódica e flutuante de dejecções diarreicas e sintomas dispépticos, sem subida dos níveis de IgM, hiperviscosidade ou citopenias.
No fim de Dezembro de 2015 foi admitida no serviço de urgência do hospital da área de residência por quadro de prostração e adinamia associado a dejecções diarreicas, vómitos e dor abdominal com 2 dias de evolução. Na admissão nesse hospital apresentava quadro de choque séptico com disfunção multiorgânica, de focalização pouco clara. Evolução desfavorável rapidamente progressiva, apesar das medidas terapêuticas instituídas, tendo culminado no óbito no próprio dia.
Revisão Teórica
A) EPIDEMIOLOGIA E PATOGÉNESE
A MW é uma doença rara, apresentando uma incidência de 3 casos/milhão/ano.2,5 A idade média de diagnóstico situa-se entre os 63 e os 68 anos, sendo incomum em idades inferiores a 40 anos.6 Esta entidade clínica manifesta uma preponderância pelo sexo masculino (cerca de 60%) e ocorre mais frequentemente em doentes de etnia caucasiana (~90% - 95%).7-8
A etiologia é desconhecida. Alguns estudos têm colocado a possibilidade de associação entre a MW e infecção por vírus da hepatite C (VHC), doenças auto-imunes ou exposição a determinados químicos (pesticidas, asbestos, etc ). Todavia, estas hipóteses estão por confirmar.9-10
Foram identificadas várias mutações somáticas e anomalias cromossómicas nas células de doentes com MW. Estas alterações parecem ocorrer numa fase tardia do processo de diferenciação celular dos linfócitos B.2,11,12 Essas células tornam-se incapazes de realizar o switch de isotipo de cadeia pesada, levando ao aparecimento de uma população clonal de linfócitos B produtores de IgM.13-18 As características imunofenotípicas na MW são variáveis. Contudo, as células tumorais expressam os marcadores de superífice e citoplasmáticos das células B (CD 19 + e CD 20 +), com ausência do CD10 e do CD23 e, em 80% - 95% dos casos, do CD5.2,13-14
A delecção do braço longo do cromossoma 6 é a única alteração citogenética identificada nos doentes com MW, até ao momento.2,13,14
A paraproteína monoclonal IgM causa lesão de órgão através de pelo menos 4 mecanismos conhecidos: deposição tecidular de IgM, infiltração de tecido/órgão por células tumorais, aumento da viscosidade sanguínea consequente à presença de IgM sérica e reacções imuno-mediadas, nas quais a IgM monoclonal se comporta como auto-anticorpo.2,15
B) MANIFESTAÇÕES CLÍNICAS
Cerca de 25% dos doentes encontram-se assintomáticos no momento do diagnóstico. Na maioria dos casos os sintomas são atribuíveis à presença da paraproteína monoclonal em circulação e/ou a infiltração de tecidos/órgãos.2,7,16 A deposição de IgM nos glomérulos, no intestino ou na pele é um dos mecanismos de lesão descritos.2
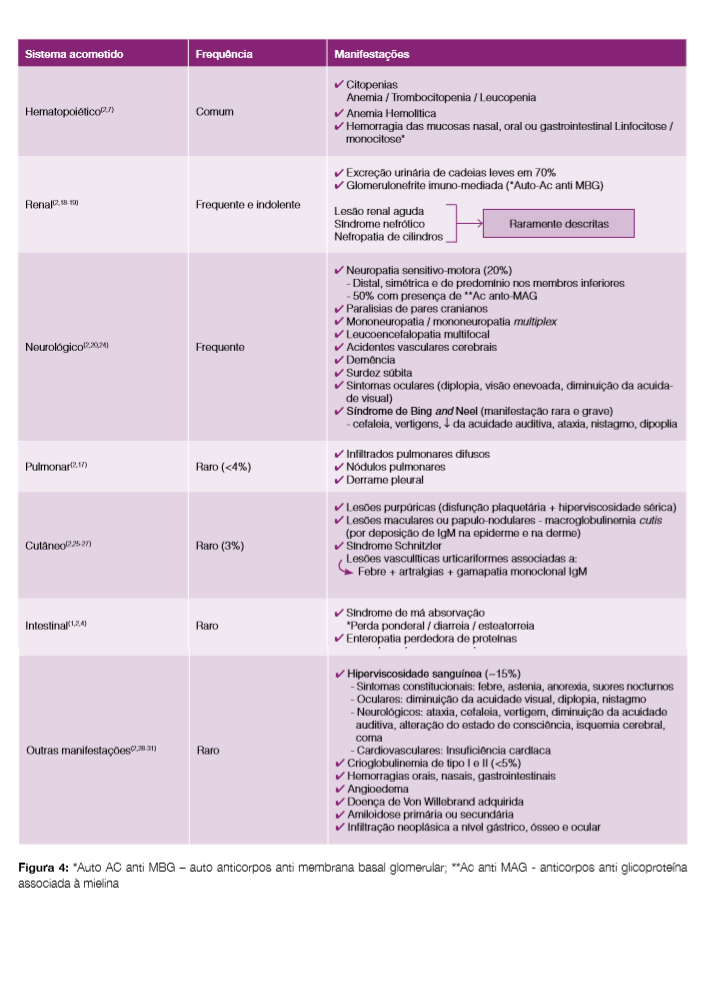
A MW é uma doença sistémica, podendo afectar praticamente todos os órgãos. Na Fig 4 encontram-se descritas as manifestações clínicas associadas a esta patologia.1,2,4,7,17-31
A anemia hemolítica auto-imune por anticorpos frios é uma das manifestações clínicas observada nos doentes com MW. Esta ocorre devido ao comportamento da IgM como auto-anticorpo, reagindo contra antigénios expressos na superfície dos glóbulos vermelhos e causando a sua destruição.2,15 No entanto, a etiologia da anemia na MW é multifactorial, não sendo a hemólise o único mecanismo implicado.7
A infiltração celular de gânglios linfáticos (25%), do fígado (24%) e do baço (19%) condiciona o desenvolvimento de organomegalias, presentes em cerca 20% - 25% dos doentes.7
O atingimento intestinal é raro,1,2,4 e deve-se essencialmente à deposição de paraproteína IgM e/ou à infiltração por células tumorais produtoras de IgM na lâmina própria da mucosa intestinal, embora possa haver contributo de outros mecanismos (nomeadamente deposição de proteína amilóide, aumento da viscosidade do fluído intestinal pela presença de IgM e sobreinfecção bacteriana ou parasitária).1 As alterações na mucosa intestinal são identificadas sobretudo no intestino delgado, sendo raros ou mesmo inexistentes os casos de atingimento do cólon. Diarreia, perda ponderal e esteatorreia são as expressões clínicas mais comuns de síndrome de má absorção.1,2,4 Para além disso, quando os quadros de má absorção são severos podem cursar com enteropatia perdedora de proteínas, entre as quais se encontram as proteínas com acção anticoagulante, com consequente aumento do risco trombótico.1,4
Nos doentes em que existe suspeita de síndrome de má absorção, a realização de estudos endoscópicos é essencial. O aspecto macroscópico da mucosa intestinal é variável, sendo a presença de edema ou de placas esbranquiçadas dois dos achados mais comuns. As alterações histológicas mais frequentemente identificadas são a linfangiectasia intestinal, a deposição de IgM monoclonal e a infiltração da mucosa por células tumorais. Pode haver deposição de substância amilóide (identificada pelo vermelho de Congo), sendo obrigatório equacionar nestes casos a amiloidose como diagnóstico alternativo ou cumulativo.
C) DIAGNÓSTICO
O dignóstico de MW implica coexistência de 3 dos critérios expressos na Tabela 1.2,32-33

D) TRATAMENTO
Actualmente, não existe um tratamento curativo para a macroglobulinemia de Waldenström,2 tornando-se necessária uma selecção criteriosa dos doentes a propor para terapêuticas mais agressivas.
Os doentes assintomáticos (forma indolente), com hemoglobina > 11 g/dL e plaquetas > 120 000/µL, representam cerca de 25% de todos os doentes com MW e não devem ser propostos para qualquer tipo de tratamento até que se desenvolvam sintomas.34 Esta conduta é baseada no resultado de vários estudos que demonstraram o bom prognóstico deste grupo de doentes, cuja sobrevida média a 10 anos ronda os 70% a 75%, próxima da população em geral.35-37 Contudo, a vigilância regular (cada 4-6 meses) não pode ser descurada, uma vez que a progressão da forma indolente para a sintomática tem maior probabilidade de ocorrer nos primeiros 5 anos após o diagnóstico, com uma taxa de progressão anual estimada entre 4% e 12%.35-37
Nos doentes sintomáticos ou com alterações analíticas como anemia (Hb <11g/dL), neutropenia ou trombocitopenia (<120 000 plaquetas/µL), a quimioterapia deve ser iniciada, tendo como objectivos o controlo de sintomas e a prevenção de lesão de órgão.2,38-40 Os sintomas podem ser divididos em três categorias: sintomas sistémicos decorrentes do processo linfoproliferativo; sintomas atribuíveis à hiperviscosidade secundária à hiperprodução de IgM; e neuropatia paraneoplásica.
A escolha do tratamento a propor deve ter em conta factores como a idade, a gravidade dos sintomas, a elegibilidade para transplante autólogo de células hematopoiéticas e as comorbilidades.2,38-40
A plasmaferese é uma opção terapêutica reservada para doentes que se apresentam com síndrome de hiperviscosidade.2,38-41 Dado que a IgM monoclonal se encontra praticamente restrita ao espaço intravascular, a plasmaferese tem a capacidade de a remover, proporcionando alívio imediato dos sintomas. No entanto, é apenas uma terapêutica sintomática, sendo necessário associar tratamento dirigido ao mecanismo de produção da proteína monoclonal (quimioterapia).2,38-41
A maioria dos peritos recomenda uma quimioterapia combinada, onde deve estar incluído o anticorpo monoclonal anti-CD20 - rituximab. Vários estudos têm demonstrado maiores taxas de resposta à terapêutica, menor toxicidade e maior sobrevida nos doentes submetidos a terapêutica com inclusão de rituximab.5,37,42
Em alguns doentes poderá ser preferida a monoterapia com rituximab (375 mg/m2 semanalmente durante 4 semanas).43-44 Nestes casos, são seleccionados doentes com sintomatologia ligeira, citopenias moderadas (hemoglobina 10-11 g/dL, plaquetas 100 000-120 000/L), neuropatia ou anemia hemolítica. O tempo médio para obter resposta parcial ao tratamento ronda os 4 meses, sendo a melhor resposta alcançada por volta dos 17 meses. Não há relatos de resposta completa. Níveis elevados de IgM podem persistir por um período superior a 4 meses e não são critério de falência terapêutica.37,45
Outra consideração importante no tratamento de doentes com MW é a elegibilidade para transplante autólogo de células estaminais. Nos doentes em que haja a perspectiva de transplante, devem ser preferidos esquemas de quimioterapia que condicionem menor toxicidade para as células estaminais. Actualmente, a maioria dos autores faz referência a três esquemas passíveis de serem utilizados nestes doentes:
1. dexametasona, rituximab e ciclofosfamida46
2. bortezomib, rituximab com ou sem associação de dexametasona47-49
3. bendamustina em associação com rituximab50
Outros esquemas possíveis no tratamento de doentes com MW incluem agentes com maior potencial de toxicidade para as células estaminais, como por exemplo esquemas combinados de talidomida ou fludarabina em associação com rituximab. Em doentes com doença refractária ou elevada taxa de recidiva, que apresentem bom estado geral, pode ser tentando o uso isolado de análogos de purina (fludarabina, cladribina) ou agentes alquilantes (clorambucilo).51-56
A esplenectomia deve ser ponderada nos doentes com dor atribuível à esplenemogalia ou com sintomatologia não controlável relacionada com o hiperesplenismo.
E) FOLLOW-UP E PROGNÓSTICO
Todos os doentes com macroglobulinemia de Waldenström devem ter seguimento médico periódico, com o objectivo de avaliar a resposta à terapêutica e de monitorizar sinais de recidiva de doença. A periodicidade de cada avaliação é determinada caso a caso.
Deve ser efectuada história clínica, exame físico e doseamento de IgM no soro ou na urina, antes de cada tratamento.
Apesar de constituir um método simples e acessível, o doseamento dos níveis de IgM deve ser interpretado com cuidado, dado que o mesmo pode apresentar flutuações independentes da actividade da doença (iatrogénicas ou dependentes do método usado).
A rápida deterioração do estado geral e da capacidade funcional aliada ao aparecimento de adenomegalias, aumento dos níveis de LDH e diminuição dos níveis de IgM, são sinais que indiciam possibilidade de transformação histológica em linfomas de alto grau.
A sobrevida média após o diagnóstico é de aproximadamente 5 anos.57-58 Porém, os doentes com MW constituem um grupo heterogéneo e raro o que conduz a uma grande variabilidade nos outcomes, impondo dificuldades na construção de um modelo de prognóstico.
Ao longo das últimas décadas, têm sido propostos vários modelos,57-61 tendo por base a utilização de múltiplas variáveis clínicas e laboratoriais. Foram identificados como factores de mau prognóstico comuns aos vários modelos a idade avançada, as citopenias e níveis elevados de ß2 microglobulina. O ISSWM é um dos modelos mais utilizados na prática clínica, contemplando cada uma das variáveis acima descritas, acrescidas do valor de IgM. Neste score os doentes são estratificados em 3 grupos: baixo risco (0 ou 1 ponto - que não corresponda à variável idade) e têm uma sobrevida estimada a 5 anos de 87%; risco Intermédio (idade ou 2 pontos) com sobrevivida estimada de 68% e Alto risco (3 ou mais pontos) com uma sobrevida estimada a 5 anos de 36%.61
Recentemente, na sequência de um estudo prospectivo multicêntrico observacional, constatou-se que a elevação de LDH sérica é também um factor de mau prognóstico, devendo ser adicionada aos scores de prognóstico pré-existentes.
Os doentes com forma indolente de MW têm prognóstico favorável sem qualquer tratamento e não estão contemplados em alguns dos modelos existentes.59-62
Assim, os actuais scores encontram-se ainda em fase de validação, necessitando de maior número de estudos prospectivos para reforçar a sua capacidade preditiva.
Comentário Final
Este caso ilustra uma manifestação rara de uma doença rara e relembra a MW como diagnóstico diferencial perante uma síndrome de má absorção.
Analisando de forma retrospectiva o caso clínico, verifica-se que as manifestações clínicas expressas em 2010 poderiam já configurar MW.
A forma de apresentação com envolvimento intestinal exclusivo é extremamente rara, existindo cerca de 25 casos descritos na literatura. No caso particular desta doente, o acometimento intestinal deve-se à deposição de proteína monoclonal na mucosa intestinal, sem evidência de infiltração neoplásica da mesma como mecanismo concomitante. Além disso, a presença de linfangiectasia, característica presente em 19 de 25 casos, não parece ter relação com os níveis de IgM em circulação, tal como foi reportado na revisão de Pratz K et al na Mayo Clinic.1
Salienta-se ainda que esta doente não apresentava quaisquer outras manifestações, como citopenias ou sintomas atribuíveis a hiperviscosidade. A adopção de medidas dietéticas possibilitou resolução da diarreia e melhoria do estado nutricional. Estes dois factos sustentaram a manutenção da abordagem mais conservadora, protelando o início de quimioterapia.
Nos casos até agora reportados de atingimento intestinal, a região do delgado é a mais frequentemente envolvida, sendo raras ou mesmo inexistentes as descrições de acometimento cólico. Esta constatação obriga a ponderar a possibilidade da existência de factores (eventualmente imunoquímicos) até agora desconhecidos que determinem a deposição preferencial da proteína monoclonal nesta localização.
Infelizmente, no final de 2015 a doente apresentou um quadro de instalação aguda e rápida evolução de choque séptico com disfunção multiorgânica, que culminou na morte. Não foi possível identificar o foco infeccioso, mas admite-se que a imunodepressão relacionada com a doença e com a esplenectomia possam ter contribuído para o desfecho desfavorável. Quanto à macroglobulinemia de Waldenström em si, a evolução estava a ser indolente, não havendo indícios que sugerissem progressão da doença (nomeadamente agravamento da anemia, aumento da frequência das dejecções diarreicas, síndrome constitucional ou mesmo alterações analíticas face ao basal da doente) nos 2 meses prévios a este episódio.
Referencias
1. Pratz K, Dingli D, Smyrk T, Lust JA. Intestinal Lymphangiectasia with protein-Losing enteropathy in Waldenstrom Macroglobulinemia. Medicine. 2007;86:210-4. [ Links ]
2. Vijay A, Gertz M. Waldenstrom macroglobulinemia. Blood. 2007; 109:5096103. [ Links ]
3. Gertz MA. Waldenstrom macroglobulinemia. Hematology. 2012;17 Suppl 1:S112-6. [ Links ]
4. Veloso F, Fraga J, Saleiro J. Macroglobulinemia and small intestinal disease. J Clin Gastrenterol. 1998; 10: 546-50. [ Links ]
5. Fonseca R, Hayman S. Waldenström macroglobulinaemia. Br J Haematol. 2007; 138:700-20. [ Links ]
6. García-Sanz R, Montoto S, Torrequebrada A, de Coca AG, Petit J, Sureda A, et al. Waldenström macroglobulinaemia: presenting features and outcome in a series with 217 cases. Br J Haematol. 2001; 115:575-82. [ Links ]
7. Benjamin M, Reddy S, Brawley OW. Myeloma and race: a review of the literature. Cancer Metastasis Rev. 2003; 22:87. [ Links ]
8. Royer RH, Koshiol J, Giambarresi TR, Vasquez LG, Pfeiffer RM, McMaster ML. Differential characteristics of Waldenström macroglobulinemia according to patterns of familial aggregation. Blood. 2010; 115:4464-71. [ Links ]
9. Linet MS, Humphrey RL, Mehl ES, Brown LM, Pottern LM, Bias WB, et al. A case-control and family study of Waldenstrom´s macroglobulinemia. Leukemia. 1993; 7:1363-9. [ Links ]
10. Kristinsson SY, Koshiol J, Björkholm M, Goldin LR, McMaster ML, Turesson I, et al. Immune-related and inflammatory conditions and risk of lymphoplasmacytic lymphoma or Waldenstrom macroglobulinemia. J Natl Cancer Inst. 2010; 102:557-67. [ Links ]
11. Aoki H, Takishita M, Kosaka M, Saito S. Frequent somatic mutations in D and/ or JH segments of Ig gene in Waldenströms macroglobulinemia and chronic lymphocytic leukemia (CLL) with Richter´s syndrome but not in common CLL. Blood. 1995; 85:1913. [ Links ]
12. Ciric B, VanKeulen V, Rodriguez M, Kyle RA, Gertz MA, Pease LR. Clonal evolution in Waldenstrom macroglobulinemia highlights functional role of B-cell receptor. Blood. 2001; 97:321-3. [ Links ]
13. Schop RF, Kuehl WM, Van Wier SA, , Ahmann GJ, Price-Troska T, Bailey RJ, et al. Waldenström macroglobulinemia neoplastic cells lack immunoglobulin heavy chain locus translocations but have frequent 6q deletions. Blood. 2002; 100:2996-3001. [ Links ]
14. Kriangkum J, Taylor BJ, Treon SP, Mant MJ, Belch AR, Pilarski LM. Clonotypic IgM V/D/J sequence analysis in Waldenstrom macroglobulinemia suggests an unusual B-cell origin and an expansion of polyclonal B cells in peripheral blood. Blood. 2004; 104:2134-42. [ Links ]
15. Stone MJ, Merlini G, Pascual V. Autoantibody activity in Waldenstrom´s macroglobulinemia. Clin Lymphoma. 2005; 5: 225-9. [ Links ]
16. Dimopoulos MA, Panayiotidis P, Moulopoulos LA, et al. Waldenström´s macroglobulinemia: clinical features, complications, and management. J Clin Oncol. 2000; 18:214-26. [ Links ]
17. Fadil A, Taylor DE. The Lung and Waldenstrom´s Macroglobulinemia. South Med J. 1998;91: 681-5. [ Links ]
18. Veltman GA, van Veen S, Kluin-Nelemans JC. Renal disease in Waldenström´s macroglobulinaemia. Nephrol Dial Transplant. 1997; 12:1256-59. [ Links ]
19. Lindström FD, Hed J, Eneström S. Renal pathology of Waldenström´s macroglobulinaemia with monoclonal antiglomerular antibodies and nephrotic syndrome. Clin Exp Immunol. 1980; 41:196. [ Links ]
20. Civit T, Coulbois S, Baylac F, Taillandier L, Auque J. Macroglobulinemie de Waldenstrom et proliferation lymphoplasmocytaire cerebrale: le syndrome de Bing et Neel. A propos d’une nouvelle observation. Neurochirurgie. 1997; 43:245-9
21. Nobile-Orazio E, Marmiroli P, Baldini L, Spagnol G, Barbieri S, Moggio M, et al. Peripheral neuropathy in macroglobulinemia: incidence and antigen-specificity of M proteins. Neurology 1987; 37:1506-14. [ Links ]
22. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 16-1999. A 71-year-old man with progressive weakness and a gammopathy. N Engl J Med. 1999; 340:1661-9. [ Links ]
23. Rudnicki SA, Harik SI, Dhodapkar M, Barlogie B, Eidelberg D. Nervous system dysfunction in Waldenström´s macroglobulinemia: response to treatment. Neurology. 1998; 51:1210-3. [ Links ]
24. Baehring JM, Hochberg EP, Raje N, Ulrickson M, Hochberg FH. Neurological manifestations of Waldenström macroglobulinemia. Nat Clin Pract Neurol. 2008; 4:547-56. [ Links ]
25. Cobb MW, Domloge-Hultsch N, Frame JN, Yancey KB. Waldenström macroglobulinemia with an IgM-kappa antiepidermal basement membrane zone antibody. Arch Dermatol. 1992; 128:372. [ Links ]
26. Lowe L, Fitzpatrick JE, Huff JC, Shanley PF, Golitz LE. Cutaneous macroglobulinosis. A case report with unique ultrastructural findings. Arch Dermatol. 1992; 128:377-80. [ Links ]
27. Gressier L, Hotz C, Lelièvre JD, Carlotti A, Buffet M, Wolkenstein P, et al. Cutaneous macroglobulinosis: a report of 2 cases. Arch Dermatol. 2010; 146:65-9. [ Links ]
28. Gertz MA, Kyle RA. Hyperviscosity syndrome. J Intensive Care Med 1995; 10:128-9. [ Links ]
29. Kwann HC, Bongu A. The hiperviscosity syndromes. Semin Thromb Hemost; 1999;25:199-208. [ Links ]
30. Michael AB, Lawes M, Kamalarajan M. Cryoglobulinaemia as an acute presentation of Waldenstrom´s macroglobulinaemia. Br J Haematol. 2004; 124:565. [ Links ]
31. Terrier B, Jaccard A, Harousseau JL, Delarue R, Tournilhac O, Hunault-Berger M, et al. The clinical spectrum of IgM-related amyloidosis: a French nationwide retrospective study of 72 patients. Medicine. 2008; 87:99-109. [ Links ]
32. Owen RG, Treon SP, Al-Katib A. Clinicopathological definition of Waldenstrom´s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom´s Macroglobulinemia. Semin Oncol. 2003; 30:110-5. [ Links ]
33. Swerdlow SH, Campo E, Harris NL, editors. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008. [ Links ]
34. Kyle RA, Treon SP, Alexanian R. Prognostic markers and criteria to initiate therapy in Waldenstrom´s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstroms Macroglobulinemia. Semin Oncol. 2003; 30:116. [ Links ]
35. Gobbi PG, Baldini L, Broglia C, Goldaniga M, Comelli M, Morel P, et al. Prognostic validation of the international classification of immunoglobulin M gammopathies: a survival advantage for patients with immunoglobulin M monoclonal gammopathy of undetermined significance? Clin Cancer Res. 2005; 11:1786-90. [ Links ]
36. Dhodapkar MV, Hoering A, Gertz MA, Rivkin S, Szymonifka J, Crowley J, et al. Long-term survival in Waldenstrom macroglobulinemia: 10-year follow-up of Southwest Oncology Group-directed intergroup trial S9003. Blood. 2009; 113:793-6. [ Links ]
37. Ansell SM, Kyle RA, Reeder CB, Fonseca R, Mikhael JR, Morice WG, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc. 2010; 85:824-33. [ Links ]
38. Johnson SA, Birchall J, Luckie C, Oscier DG, Owen RG; Haemato-Oncology Task Force of the British Committee for Standards in Haematology. Guidelines on the management of Waldenström macroglobulinaemia. Br J Haematol 2006; 132:683-97. [ Links ]
39. Gertz MA. Waldenström macroglobulinemia: a review of therapy. Am J Hematol. 2005; 79:147. [ Links ]
40. Treon SP, Gertz MA, Dimopoulos M. Update on treatment recommendations from the Third International Workshop on Waldenstrom´s macroglobulinemia. Blood. 2006; 107:3442. [ Links ]
41. Szczepiorkowski ZM, Winters JL, Bandarenko N. Guidelines on the use of therapeutic apheresis in clinical practice--evidence-based approach from the Apheresis Applications Committee of the American Society for Apheresis. J Clin Apher. 2010; 25:83. [ Links ]
42. Dimopoulos MA, Gertz MA, Kastritis E. Update on treatment recommendations from the Fourth International Workshop on Waldenstrom´s Macroglobulinemia. J Clin Oncol. 2009; 27:120. [ Links ]
43. Dimopoulos MA, Alexanian R, Gika D, Tournilhac O, Leblond V, Morel P. Treatment of Waldenstrom´s macroglobulinemia with rituximab: prognostic factors for response and progression. Leuk Lymphoma 2004; 45:2057-8. [ Links ]
44. Gertz MA, Rue M, Blood E, Kaminer LS, Vesole DH, Greipp PR. Multicenter phase 2 trial of rituximab for Waldenström macroglobulinemia (WM): an Eastern Cooperative Oncology Group Study (E3A98). Leuk Lymphoma 2004; 45:2047-55. [ Links ]
45. Treon SP, Branagan AR, Hunter Z, Santos D, Tournhilac O, Anderson KC. Paradoxical increases in serum IgM and viscosity levels following rituximab in Waldenstrom´s macroglobulinemia. Ann Oncol. 2004; 15:1481-3. [ Links ]
46. Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, Zervas K, Tsatalas C, Kokkinis G, et al. Primary treatment of Waldenström macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol. 2007; 25:3344-9. [ Links ]
47. Treon SP, Ioakimidis L, Soumerai JD, Patterson CJ, Sheehy P, Nelson M, et al. Primary therapy of Waldenström macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Oncol. 2009; 27:3830-5. [ Links ]
48. Ghobrial IM, Xie W, Padmanabhan S, Badros A, Rourke M, Leduc R, et al. Phase II trial of weekly bortezomib in combination with rituximab in untreated patients with Waldenström macroglobulinemia. Am J Hematol. 2010; 85:670-4. [ Links ]
49. Agathocleous A, Rohatiner A, Rule S, Hunter H, Kerr JP, Neeson SM, et al. Weekly versus twice weekly bortezomib given in conjunction with rituximab, in patients with recurrent follicular lymphoma, mantle cell lymphoma and Waldenström macroglobulinaemia. Br J Haematol. 2010; 151:346-53. [ Links ]
50. Steven TP. How I treat Waldenstr?m macroglobulinemia. Blood. 2015; 126: 721-32 [ Links ]
51. Treon SP, Soumerai JD, Branagan AR, Hunter ZR, Patterson CJ, Ioakimidis L, et al. Thalidomide and rituximab in Waldenstrom macroglobulinemia. Blood. 2008; 112:4452-7. [ Links ]
52. Leblond V, Johnson S, Chevret S, Copplestone A, Rule S, Tournilhac O, et al. Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenström macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J Clin Oncol. 2013; 31:301-7. [ Links ]
53. Leleu X, Soumerai J, Roccaro A, Hatjiharissi E, Hunter ZR, Manning R, et al. Increased incidence of transformation and myelodysplasia / acute leucemia in patients with Waldenstrom macroglobulinemia treated with nucleoside analogs. J Clin Oncol. 2009; 27:250-5. [ Links ]
54. Yang K, Tan J, Wu T. Alkylating agents for Waldenstrom´s macroglobulinaemia. Cochrane Database Syst Rev. 2009; CD006719. [ Links ]
55. Anagnostopoulos A, Hari PN, Pérez WS, Ballen K, Bashey A, Bredeson CN, et al. Autologous or allogeneic stem cell transplantation in patients with Waldenstrom´s macroglobulinemia. Biol Blood Marrow Transplant 2006; 12:84554. [ Links ]
56. Garnier A, Robin M, Larosa F, Golmard JL, Le Gouill S, Coiteux V, et al. Allogeneic hematopoietic stem cell transplantation allows long-term complete remission and curability in high-risk Waldenström’s macroglobulinemia. Results of a retrospective analysis of the Société Française de Greffe de Moelle et de Thérapie Cellulaire. Haematologica. 2010; 95:950-5.
57. Sekhar J, Sanfilippo K, Zhang Q, Trinkaus K, Vij R, Morgensztern D. Waldenström macroglobulinemia: a surveillance, epidemiology, and end results database review from 1988 to 2005. Leuk Lymphoma. 2012; 53:1625-6. [ Links ]
58. Kristinsson SY, Eloranta S, Dickman PW, Andersson TM, Turesson I, Landgren O, et al. Patterns of survival in lymphoplasmacytic lymphoma/Waldenström macroglobulinemia: a population-based study of 1,555 patients diagnosed in Sweden from 1980 to 2005. Am J Hematol. 2013; 88:60-5. [ Links ]
59. Merlini G, Baldini L, Broglia C, Comelli M, Goldaniga M, Palladini G, et al. Prognostic factors in symptomatic Waldenstrom´s macroglobulinemia. Semin Oncol. 2003; 30:211-5. [ Links ]
60. Ghobrial IM, Fonseca R, Gertz MA, Plevak MF, Larson DR, Therneau TM, et al. Prognostic model for disease-specific and overall mortality in newly diagnosed symptomatic patients with Waldenstrom macroglobulinaemia. Br J Haematol 2006; 133:158-64. [ Links ]
61. Morel P, Duhamel A, Gobbi P, Dimopoulos MA, Dhodapkar MV, McCoy J, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood. 2009; 113:4163-70. [ Links ]
62. Kastritis E, Kyrtsonis M, Hadjiiharissi E, Symeonidis A, Michalis E, Repoussis P, et al. Validation of the International Prognostic Scoring System (IPSS) for Waldenstrom´s macroglobulinemia (WM) and the importance of serum lactate dehydrogenase (LDH). Leuk Res. 2010;34:1340-3. [ Links ]
Correspondência: Nídia Pereira nidiapereiraoliveira@gmail.com
Serviço de Medicina Interna, Hospital Pedro Hispano, Matosinhos, Portugal
Rua Dr. Eduardo Torres, 4464-513, Senhora da Hora
Conflitos de Interesse: Os autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
Conflicts of interest: The authors have no conflicts of interest to declare.
Fontes de Financiamento: Não existiram fontes externas de financiamento para a realização deste artigo.
Financing Support: This work has not received any contribution, grant or scholarship.
Direito à Privacidade e Consentimento Informado: Os autores declaram que nenhum dado que permita a identificação do doente aparece neste artigo.
Confidentiality of data: The authors declare that they have followed the protocols of their work center on the publication of data from patients.
Proteção de Seres Humanos e Animais: Os autores declaram que não foram realizadas experiências em seres humanos ou animais.
Protection of human and animal subjects: The authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Recebido: 06/01/2017
Aceite: 10/03/2017


{kind=link}