








Servicios Personalizados
Revista
Articulo
 Portugués (pdf)
Portugués (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicina Interna
versión impresa ISSN 0872-671X
Medicina Interna vol.25 no.1 Lisboa mar. 2018
https://doi.org/10.24950/rspmi/CC/146/1/2018
CASOS CLÍNICOS / CASE REPORTS
Angioedema Hereditário: A Importância da Suspeita Clínica e do Tratamento Adequado
Hereditary Angioedema: The Importance of Clinical Suspicion and Proper Treatment
Ana Araújo1, Joel Pinto1, Paulo Almeida1, Beatriz Pinheiro1, Ana Morete2
1Serviço de Medicina Interna, Centro Hospitalar do Baixo Vouga, Aveiro, Portugal
2Serviço de Imunolaergologia, Centro Hospitalar do Baixo Vouga, Aveiro, Portugal
RESUMO
O angioedema hereditário (AEH) é uma síndrome caracterizada por episódios recorrentes de edema submucoso e/ou subcutâneo, associados à deficiência quantitativa ou funcional do inibidor C1-esterase (C1-inib). Os autores apresentam o caso de um homem, de 40 anos, com história familiar de AEH. Apresentava crises recorrentes de dor abdominal, náuseas e vómitos. O estudo imunológico revelou níveis diminuídos de C1-inib e C4 e os de C1q eram normais. Excluiu-se angioedema adquirido, diagnosticando-se AEH tipo 1. Recorreu várias vezes ao serviço de urgência por dor abdominal intensa, com abdómen distendido, difusamente doloroso. Contudo o estudo analítico e imagiológico não apresentavam alterações. Foi administrada injecção subcutânea de icatibant, com resolução do quadro. O AEH é uma doença rara, potencialmente fatal, nomedamente durante as crises. É fundamental o diagnóstico atempado e terapêutica adequada.
Palavras-chave:Angioedema Hereditário; Icatibant; Proteína Inibidora do Complemento C1.
ABSTRACT
Hereditary angioedema (HAE) is a syndrome characterized by episodic swelling of subcutaneous and submucosal tissues, due to a deficiency or dysfunctionality in functional C1 esterase inhibitor (C1-inh). The authors present a case of a 40 year-old man with a positive family history of HAE, recurrent episodes of abdominal pain, nausea and vomiting. The immunological study revealed low levels of C1-inh and C4 and normal levels of C1q. Acquired angioedema was excluded, making the diagnosis of type 1 HAE. The patient presented several times to the emergency department with severe abdominal pain, diffuse abdominal tenderness with guarding. However, laboratory and imagiologic studies had no alterations. A subcutaneous injection of icatibant was administered with resolution of symptoms. HAE is a rare and potentially fatal disease, mainly during acute attacks. An early diagnosis and a proper treatment are fundamental.
Keywords: Angioedemas, Hereditary; Complement C1 Inhibitor Protein; Icatibant.
Introdução
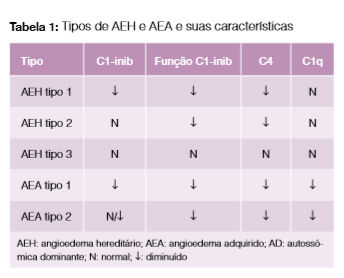
O angioedema hereditário (AEH) é uma doença genética, autossómica dominante, atribuída à deficiência de C1-inib, causada pela mutação no gene que o codifica, localizado no cromossoma 11.1 Afecta entre 1:10 000 e 1:50 000 indivíduos, embora a sua raridade possa subestimar a prevalência real.2 Mutações espontâneas ocorrem em cerca de 25% dos doentes sem história familiar de AEH.3 Estão descritos três tipos de AEH: tipo 1 (cerca de 85%), caracterizado por deficiência quantitativa e funcional de C1-inib; tipo 2 (cerca de 15%), em que existe deficiência funcional, sem défice quantitativo e tipo 3, o mais raro, sem alterações quantitativas ou da função de C1-inib4 (Tabela 1). O C1-inh é uma a2-globulina plasmática da família dos inibidores de proteases serínicas, produzido nos hepatócitos, sendo a sua principal função biológica a prevenção de um aumento da permeabilidade vascular, regulando o sistema de síntese das cininas e da via clássica da activação do complemento. A deficiência de C1-inib desencadeia uma activação espontânea e excessiva do sistema das cininas e do complemento. Os mediadores libertados (bradicinina e C2) são os responsáveis pelo aumento da permeabilidade vascular, saída do plasma e aparecimento do angioedema.2,5-6 Os sintomas começam na primeira ou segunda década, podem agravar durante a puberdade e persistem toda a vida.5 A expressão clínica é altamente variável, desde casos assintomáticos até episódios quase fatais. As crises podem ser precipitadas por trauma minor, stress emocional, exposição ao frio, alimentos e fármacos (contracetivos orais, inibidores da enzima de conversão de angiotensina (IECA) e antagonistas dos receptores de angiotensina). Clinicamente caracteriza-se por episódios recorrentes de edema subcutâneo e submucoso, localizado, não pruriginoso, não depressível, com envolvimento das extremidades, genitais, sistema digestivo, face e vias respiratórias.2,5,6 As crises podem ser precedidas de rash não pruriginoso, com agravamento sintomático nas primeiras 24 horas, e resolução às 48-72 horas.5 O diagnóstico definitivo é laboratorial. Uma vez que os níveis de C4 estão virtualmente baixos em todos os doentes com AEH, torna-se um bom teste de rastreio. Deve determinar-se o C1-inib quantitativo e funcional, fazendo o diagnóstico diferencial entre AEH tipo 1 e 2.2,5 O estudo imunológico pode ser normal no caso do AEH tipo 3.4 O tratamento assenta em três pilares: abordagem da crise aguda, profilaxia a curto prazo e a longo prazo.7 Os anti-histaminicos, corticóides e adrenalina são opções terapêuticas ineficazes.8
Caso Clínico
Doente de 40 anos, do sexo masculino, raça caucasiana, sem contexto epidemiológico relevante e sem medicação crónica. Referia quadro com início aos 25 anos, de dor abdominal ocasionalmente intensa, associado a náuseas e vómitos, com duração de 3 dias, a condicionar limitação para as actividades da vida diária. Negava alterações do trânsito intestinal, presença de sangue ou muco nas fezes, alterações cutâneas, artralgias ou febre. Alguns desses episódios motivaram recurso ao serviço de urgência, sendo medicado com antiespasmódicos. Nessas alturas não apresentava alterações analíticas e a radiografia e ecografia abdominais eram normais. Reconhecia associação destes episódios com stress emocional, sem outros factores desencadeantes. Dos antecedentes familiares referiu uma irmã com episódios de angioedema facial e pescoço e o pai com episódios de edema das extremidades autolimitados. Perante a história familiar e episódios recorrentes de dor abdominal sem causa orgânica, procedeu-se ao estudo de angioedema bradicinérgico em consulta de Imunoalergologia. O estudo analítico não apresentava alterações no hemograma, fórmula leucocitária ou plaquetas. A função renal, hepática e tiroideia eram normais, assim como a creatina quinase, mioglobina, velocidade de sedimentação e proteína-C-reactiva. A electroforese proteica e doseamento de imunoglobulinas não tinham alterações. O estudo de auto-imunidade foi negativo, assim como as serologias víricas. O VDRL foi não reactivo e o exame sumário de urina era normal. O estudo imagiológico torácico e abdominal era normal. Os testes cutâneos por picada a aeroalergénios e alimentos foram negativos. O estudo imunológico mostrou níveis diminuídos de C4 e C1-inib, com função, também diminuída, com níveis normais de C1q, compatível com o diagnóstico de AEH tipo 1. O doente não tinha descendentes. Nessa altura, foi-lhe fornecido protocolo de atuação nas crises de AEH, com fármacos e dosagens a ser utilizadas. Mantinha episódios de dor abdominal mode rada a intensa, associados a náuseas e vómitos (mais que um episódio mensal), alguns dos quais com necessidade de recurso a urgência hospitalar. Pelas características do episódio foi sempre avaliado pela cirurgia geral, apresentando taquicardia, apirexia e abdómen difusamente doloroso, com defesa, sem massas ou organomegalias palpáveis. Analiticamente e imagiologicamente sem alterações agudas. Foi sempre pedida a colaboração da Medicina Interna, que administrou icatibant, com resolução dos sintomas após 2 horas. Por apresentar mais de uma crise com envolvimento gastrointestinal foi iniciada profilaxia a longo prazo com estanozolol 2 mg diários, apresentando menos de dois episódios anuais de dor abdominal. Manteve vigilância em consulta, sem evidência de complicações da terapêutica androgénica.
Discussão e Conclusão
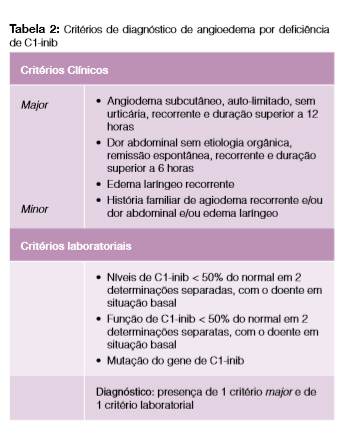
O angioedema pode ser histaminérgico, secundário à activação dos mastócitos e consequente libertação de histamina ou bradicinérgico sendo mediado pela bradicinina. Este último pode ser hereditário, adquirido, induzido por IECA ou idiopático. O nosso doente não apresentava urticária, sendo excluído angioedema histaminérgico. Nunca foi medicado com IECA, afastando a hipótese de angioedema induzido por estes fármacos. Perante os sintomas de dor abdominal recorrente e história familiar, colocou-se a hipótese de AEH. Realizou-se estudo complementar exaustivo, excluindo-se doenças auto-imunes, neoplasias, infeções víricas e bacterianas e patologia tiroideia, possíveis causas de angioedema adquirido.9 O estudo imunológico revelou níveis diminuídos de C4 e C1-inib (< 50%), com função diminuída e C1q normais, em duas determinações separadas e não em crise. A determinação de C1q é importante quando se suspeita de angioedema adquirido.6 O doente reunia critérios de diagnóstico de AEH tipo 1 (Tabela 2). Apenas foi objectivado como factor precipitante o stress.2 O estudo genético não é necessário para confirmar o diagnóstico de AEH, sobretudo com história familiar positiva.4 As queixas abdominas mimetizam quadros de abdómen agudo, sendo frequentes nestes doentes cirurgias exploratórias brancas. No caso apresentado os episódios abdominais foram sempre interpretados como gastroenterites, evitando intervenções desnecessárias. A partir do momento do diagnóstico o doente passou a ser portador de informação clínica sobre AEH, que foi fundamental para evitar procedimentos desnecessários. A ecografia abdominal é um exame complementar útil, principalmente nas crises agudas, como descrito.10 A profilaxia a longo prazo tem como objectivo reduzir o número e gravidade das crises e está indicada quando o doente tem mais que um episódio grave por mês ou um episódio potencialmente fatal. Os fármacos usados são os androgénios (danazol e estanozolol), que aumentam a produção de C1-inh e os antifibrinolíticos (ácido e-aminocapróico e ácido tranexâmico), pelacapacidade de inativação da plasmina.3,4,10 A profilaxia a curto prazo está indicada em procedimentos cirúrgicos, dentários ou invasivos programados. Nas manipulações minor está indicada a profilaxia com androgénios ou antifibrinolíticos, se manipulações major com necessidade de entubação orotraqueal estão recomendados o concentrado de C1-inib ou o Icatibant. Nas crises agudas com envolvimento laríngeo ou abdominal grave podem ser usados o concentrado do C1 –inib ou o Icatibant, sendo este o tratamento de eleição, pela comodidade de aplicação (injecção subcutânea), rápido antagonismo dos receptores da bradicinina e rápida reposição dos níveis de C1-inib.3,4,10 Se estes fármacos não estiverem disponíveis, o plasma fresco congelado, ácido tranexâmico e androgénios são uma opção.4 O doente iniciou estanozolol, que manteve na dose mínima eficaz durante 3 meses, com diminuição das crises agudas e subida dos níveis de C1inib. O Icatibant é um antagonista selectivo dos receptores ß2 da bradicinina, revertendo o aumento da permeabilidade vascular.10 Foi utilizado nas crises agudas do nosso doente, a partir do diagnóstico, com resolução das mesmas. O atraso diagnóstico continua a ser um problema importante (atraso médio de 13 a 21 anos),8 associando-se a degradação da qualidade de vida, procedimentos médicos desnecessários e demora na instituição de tratamento. O doente manteve seguimento em consulta, apresentando menos de 2 crises abdominais anuais, sem evidência de efeitos adversos relacionados com a terapêutica. É importante a abordagem multidisciplinar, pois as crises agudas implicam sempre uma observação em urgência hospitalar, sendo necessário identificar esta imunodeficiência do complemento e instituir rapidamente o tratamento adequado.
Referencias
1. Viegas LP, Ferreira MB, Santos AS, Barbosa MP. Angioedema hereditário: Experiência com icatibant em crises graves. Rev Port Imunoalergol. 2012;20:128-38. [ Links ]
2. Lumry WR. Overview of epidemiology, pathophysiology, and disease progression in hereditary angioedema. Am J Manag Care. 2013;19:103-10. [ Links ]
3. Gompels MM, Lock RJ, Abinun M, Bethune CA, Davies G, Grattan C, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol. 2005;139:379-94. [ Links ]
4. Bowen T, Cicardi M, Farkas H, Bork K, Longhurst HJ, Zuraw B, et al. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema.Allergy Asthma Clin Immunol. 2010 ;6:24. [ Links ]
5. Zuraw BL. Hereditary Angioedema. N Engl J Med. 2008;359:1027-36. [ Links ]
6. UC N, E F, WJ T. Hereditary angioedema: a broad review for clinicians. Arch Intern Med. 2001;161:2417-29. [ Links ]
7. Cicardi M, Zingale L. How do we treat patients with hereditary angioedema. Transfus Apher Sci. 2003;29:221-7. [ Links ]
8. Katelaris C, Smith W, Wong M, Jordan A. Position paper on hereditary angioedema (HAE). Australas Soc Clin Immunol Allergy. 2017:1-44. [ Links ]
9. Cadinha S, Castel-Branco MdG, Malheiro D, Lopes I. Protocolo de diagnóstico, tratamento e seguimento de doentes com angioedema hereditário. Rev Port Imunoalergol. 2005;13:377-93. [ Links ]
10. Agostoni A, Aygoren-Pursun E, Binkley KE, Blanch A, Bork K, Bouillet L, et al. Hereditary and acquired angioedema: Problems and progress: Proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004;114:51-131. [ Links ]
Correspondência: Ana Araújo anarafa.araujo@gmail.com
Serviço de Medicina Interna, Centro Hospitalar do Baixo Vouga, Aveiro, Portugal
Av. Artur Ravara - 3810-501 Aveiro
Conflitos de Interesse: Os autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
Fontes de Financiamento: Não existiram fontes externas de financiamento para a realização deste artigo.
Direito à Privacidade e Consentimento Informado: Os autores declaram que nenhum dado que permita a identificação do doente aparece neste artigo.
Protecção de Seres Humanos e Animais: Os autores declaram quenão foram realizadas experiências em seres humanos ou animais.
Recebido: 24/07/2017
Aceite: 02/10/2017