










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkMedicina Interna
versão impressa ISSN 0872-671X
Medicina Interna vol.24 no.2 Lisboa jun. 2017
ARTIGOS DE REVISÃO / REVIEW ARTICLES
Manifestações Cutâneas Paraneoplásicas de Doenças Hematológicas Oncológicas
Cutaneous Paraneoplastic Manifestations of Hematological Malignancies
Joana Azevedo Duarte1, Ana Marcos-Pinto2, João Borges-Costa2,3
1Serviço de Medicina IV, Hospital Professor Doutor Fernando da Fonseca, Lisboa, Portugal
2Serviço de Dermatologia, Hospital de Santa Maria, Centro Hospitalar Lisboa Norte, Lisboa, Portugal
4Faculdade de Medicina da Universidade de Lisboa, Lisboa, Portugal
Resumo:
A pele, enquanto maior órgão do corpo humano, pode possuir um papel fundamental no diagnóstico precoce de doenças sistémicas. As manifestações cutâneas podem ser a primeira manifestação de doenças hematológicas oncológicas (linfoma de Hodgkin, leucemia linfocítica crónica, leucemia mielóide aguda), podendo estas ser classificadas em específicas, quando são causadas pela infiltração de células malignas na pele ou pela expansão loco-regional da doença tumoral; ou inespecíficas, pela desregulação imune que ocorre nos doentes oncológicos, (caracterizadas pela ausência de células tumorais). As lesões inespecíficas são mais frequentes, têm características polimórficas e podem preceder, ser concomitantes ou ocorrer após o diagnóstico da neoplasia oculta. Neste artigo serão descritas algumas das doenças cutâneas mais frequentemente associadas a neoplasias hematológicas; nomeadamente, dermatoses neutrofílicas (síndrome de Sweet, pioderma gangrenoso e policondrite recidivante), doenças do tecido conjuntivo (dermatomiosite), prurido de etiologia desconhecida e doenças vesico-bolhosas (pênfigo paraneoplásico e vulgar). Serão ainda abordadas outras menos frequentes mas cuja associação a neoplasias está bem descrita: amiloidose, eritemas reactivos, dermatoses vasculares (livedo reticularis, eritromelalgia e vasculites) e outras (acanthosis nigricans, sinal de Leser-Trélat e ictiose adquirida). O principal objectivo desta revisão é o de salientar o importante papel de certas dermatoses que podem ser a manifestação inicial de doenças hematológicas oncológicas, levando a um diagnóstico diferencial mais direccionado.
Palavras-chave:Doenças da Pele; Neoplasias Hematológicas; Síndromes Paraneoplásicos.
Abstract
The skin, as the largest organ of the human body, can have a key role in the early diagnosis of systemic diseases. Cutaneous manifestations can be the initial symptom of certain malignant hematological diseases (Hodgkin lymphoma, chronic lymphocytic leukemia, acute myeloid leukemia). These can be classified as specific when caused by direct invasion of malignant cells in the skin or by loco-regional expansion of the tumoral disease or unspecific when caused by the altered regulation of the immune system typical of oncologic patients (characterized by absence of neoplastic cells in the skin). The unspecific injuries are more frequent, have polymorphous appearance and may precede, follow or have a parallel course with the underlying malignancy. In this article, the most frequent cutaneous diseases associated with hematological malignancies are reviewed, namely neutrophilic dermatoses (Sweet syndrome and pyoderma gangrenosum), connective tissue diseases (dermatomyositis), pruritus of unknown origin and vesiculobullous disorders (pemphigus vulgaris and paraneoplastic pemphigus). Although less common, other diseases are also associated with malignancy such as amyloidosis, reactive erythemas, vascular dermatoses (livedo reticularis, erythromelalgia and vasculitis) and other disorders (acanthosis nigricans, sign of Leser-Trélat and acquired ichthyosis). The main purpose of this review is to highlight the relevance of cutaneous manifestations that can be the first symptom of hematological malignancies and lead to differential diagnosis.
Keywords:Hematologic Neoplasms; Paraneoplastic Syndromes; Skin Diseases.
Introduction
Paraneoplastic dermatoses are a group of skin disorders that are associated to an underlying neoplasm, regardless of their distance from it.1 They exhibit a variable morphology, pathology and etiology, being their link to the internal malignancy incompletely understood. Some theories support the role of cytokine production from the tumor and its accessory cells, whilst others relay on the production of autoantibodies by the cancer cells as a cause of the paraneoplastic syndromes.2 Clinically, these manifestations can precede, follow or have a parallel course with the underlying malignancy. Even though they are not specific of cancer, they alert the doctor to conduct a detailed investigation towards excluding a neoplastic etiology. In this review we will focus on the most frequent paraneoplastic cutaneous manifestations as well as their pathophysiology, differential diagnosis and prognostic relevance.
Table 1 summarizes the different cutaneous manifestations of hematologic diseases.
Neutrophilic Dermatoses
This is a group of disorders that includes Sweet syndrome and pyoderma gangrenosum and is characterized by an intense dermal inflammatory infiltrate composed of neutrophils with little evidence of vasculitis.3
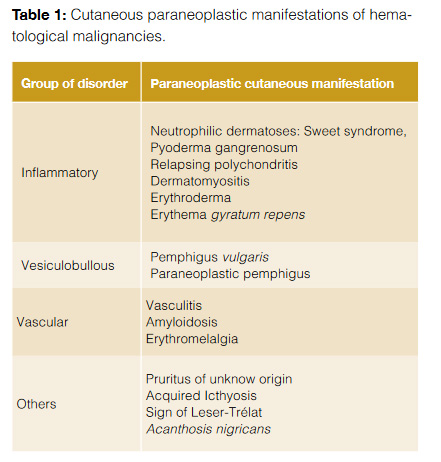
Sweet syndrome (SS) was initially described in 1964 by Dr. Robert Sweet as an acute febrile dermatosis, being characterized by the presence of fever, leukocytosis with neutrophilia and painful erythematous papules, nodules and plaques involving the limb extremities and face (Fig 1). Histologically, they show a characteristic diffuse neutrophilic infiltrate in the upper dermis.4 SS can be classified upon its clinical setting into: classical or idiopathic, malignancy-associated and drug-induced.4
Classical SS typically occurs in women aged between 30 and 50 years old and it is frequently preceded by an upper respiratory tract infection, inflammatory bowel disease or pregnancy.5 Malignancy-associated SS can occur as a paraneoplastic syndrome and it may be the first symptom of an underlying malignancy. Approximately 20% of the patients with SS have an associated cancer (being acute myelogenous leukemia the most common one) and in 40% of these, the tumor is diagnosed one month after the appearance of the skin lesions.2 It occurs more frequently in men and its recurrence is usually associated with the diagnosis or recurrence of the malignancy. Drug-induced SS most commonly occurs in patients taking granulocyte-colony stimulating factors, antibiotics, anti-epileptics, diuretics and oral contraceptives, among others.
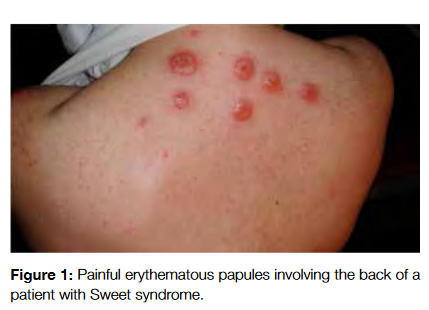
Pyoderma gangrenosum (PG) occurs as tender erythematous or violaceous nodules with tendency to ulcerate and involve large body areas occurring in the lower extremities, buttocks, abdomen and face (Fig 2). It shows endothelial swelling and fibrinoid necrosis with a predominance of leukocytes and heals leaving atrophic scars. It is estimated that 50% of cases of PG are associated with a systemic disorder (Crohns disease, ulcerative colitis), being associated with malignancy in 7% of the cases (acute myelogenous leukemia and multiple myeloma are the most common ones).2,3
The differential diagnosis of neutrophilic dermatoses includes cellulitis, lymphangitis, panniculitis, syphilis, tuberculosis, leukemia cutis, urticaria, Behçets disease, lupus erythematosus, dermatomyositis and erythema nodosum.5
Systemic corticosteroids are the therapeutic gold standard; in those patients with absolute contraindication to these drugs or active infections, potassium iodide or colchicine can be used instead (in SS). The lesions may solve spontaneously or undergo a relapsing and remitting course (up to 33% of recurrences in classical SS),5 being the prognosis usually the same as the underlying disease.
Relapsing polychondritis (RP) is an inflammatory condition affecting cartilaginous tissue, mainly in the ear, nose and tracheobronchial cartilage. It occurs in all age groups and affects men and women equally. One of the most common presenting features is acute painful swelling and redness of the external ear, which occurs in up to 40% of patients.6
The currently accepted diagnostic criteria were described by McAdamet et al7 and are1: recurrent chondritis of both auricles ; inflammatory arthritis ; chondritis of nasal cartilage ; ocular inflammation and chondritis of upper respiratory tract, cochlear and/ or vestibular. In order to diagnose RP, three or more of the above need to be present, or one plus histological diagnosis, or chondritis in two separate locations with good response to corticosteroids.7 Over 30% of patients have an associated disorder, usually autoimmune or hematologic.6 The autoimmune disorders associated are systemic vasculitis, systemic lupus erythematosus, Sjögren syndrome, overlap connective tissue disorders, rheumatoid arthritis, spondyloarthropathies and psoriasis.6 Regarding the hematologic diseases, they include myelodysplastic syndromes and Hodgkins disease.6,7 The occurrence of RP together with myelodysplasia is a marker of poor prognosis.7
The treatment is based on non-steroidal anti-inflammatory drugs in patients with mild disease and systemic corticosteroids in those with pulmonary or renal involvement.6
Connective Tissue Diseases
In this section we will focus on dermatomyosistis, the connective tissue disorder with cutaneous manifestations most frequently associated with hematological malignancies. The connective tissue diseases are a group of idiopathic inflammatory myopathies with an autoimmune etiology characterized by progressive and symmetrical weakness of proximal muscles and specific cutaneous lesions. Dermatomyositis (DM) belongs to a group of idiopathic inflammatory myopathies with an autoimmune etiology characterized by specific cutaneous lesions.8 Its first association with malignancy was described in 1916, in a patient with DM and stomach adenocarcinoma.8 Several postulates have been proposed regarding the mechanisms of association between DM and malignancy; namely the mimicry with endothelial antigens, cross-reactions between skin and muscle antigens and chronic immune stimulation (typical of DM), that would cause the activation of B and T cells with consequent malignant transformation of lymphocytes.9
The diagnosis of DM is based on Bohan and Peters criteria: symmetrical muscle weakness progressing over weeks to months; typical histological findings on muscle; elevation in serum muscle enzymes (creatine kinase, aldolase, lactate dehydrogenase); myopathic changes in electromyography and characteristic cutaneous features.9 Concerning the clinical features, they are an heliotrope rash (blue-purple discoloration of the upper eyelids) associated with local edema, V sign (an erythematous rash on the face, neck and anterior chest) or back and shoulders (shawl sign) and Gottrons papules (raised violaceous rash in the knuckles).10 Dysphagia and respiratory muscle involvement are more frequent in the cancer group, being respiratory insufficiency a frequent cause of death.8
The most frequent types of hematological malignancies related to DM are B-cell lymphoma (65% of cases), T-cell lymphoma (12.5%), Hodgkins disease, multiple myeloma, myelodysplastic syndromes, hairy cell leukemia and acute lymphocytic leukemia.9 In approximately 50% of the patients, DM is diagnosed before the hematological malignancy, with a mean interval onset of less than two years. Compared to patients with non-malignancy associated DM, these ones are older (mean age of 57 years old) and frequently of the female gender. Also, they less frequently exhibit joint involvement and anti-Jo1 antibody.9
The differential diagnosis of dermatomyositis includes systemic lupus erythematosus, systemic sclerosis, psoriasis, airborne or contact dermatitis, atopic dermatitis, photodrug eruption and cutaneous T-cell lymphoma.11
Regarding the treatment, prednisone is the first option, followed by corticosteroid-sparing drugs (azathioprine, cyclosporine). The clinical course is dependent on the underlying malignancy, as it is proven by the improvement on DM clinical features after initiation of anti-tumoral treatment. Due to its close association to malignancies, particularly hematologic ones, when DM is diagnosed, a systemic tumor investigation should be performed.
Pruritus of Unknown Origin
Pruritus is the most common symptom in dermatology and it can occur in very distinct clinical settings. It is defined as an unpleasant sensation provoking the desire to scratch.12 It results from the activation of the sensory nervous system involving the peripheral nervous system, dorsal root ganglia, spinal cord and brainstem. It can be proprioceptive (if its origin is in the skin due to inflammation or dryness), neuropathic (pathologically located along the afferent pathway), neurogenic (originates centrally but without evidence of neurologic pathology) or psychogenic. It can be further divided into chronic (lasting for more than six weeks), intractable, localized or generalized.
Paraneoplastic itch occurs before or during the diagnosis of the malignancy, it is not caused by tumoral invasion or extrinsic compression and its course is dependent on cancer therapy.13 It is associated with hematological disorders, namely Hodgkins disease (up to 30% of cases),13 multiple myeloma, lymphoma and leukemia.
When associated to Hodgkins disease (HD), it may precede the diagnosis of the disease up to five years and it is typically worse at night, starting in the legs and then spreading proximally. When generalized, it is more common in nodular sclerosis type of HD and, when localized, it usually occurs in the areas drained by lymphatic vessels invaded by the tumor. Grovers disease is an extremely pruritic papulovesicular disease that occurs in the trunk of adult men, being associated with hematological malignancies (even though it is not clear whether its association with malignancy is caused by the cutaneous effect of fever and night sweats typical seen with these conditions).13 It has been considered a B symptom in the 60s, due to its non-negligible contribution to the patients quality of life; however, a few years later it was replaced by weight loss.14 Also worth mentioning is pruritus as a feature of dermatomyositis (up to 38% of cases),12 which is itself associated to malignancy.
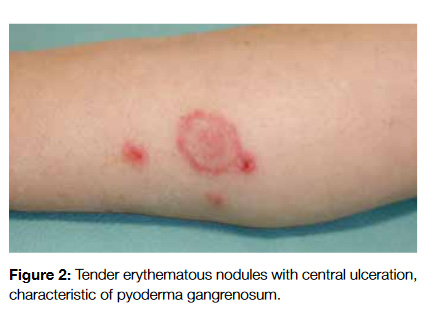
The secondary skin changes of pruritus include excoriations, lichenification, post-inflammatory hyperpigmentation, shiny finger nails, prurigo nodules and butterfly sign (areas that the patients are not able to scratch in the upper back and present a different coloration from the remaining skin) (Fig 3).12 The diagnosis can be performed by noticing these changes and asking the patient about its evolution during the day (malignancy associated pruritus usually occurs at night and impairs sleep while psychogenic one does not).
The differential diagnosis involves all the conditions that may potentially cause pruritus as a major symptom; namely infestations (scabies, pediculosis), inflammatory disorders (atopic or contact dermatitis, psoriasis, lichen planus, urticaria, drug eruptions, mastocytosis), autoimmune connective tissue disorders (lichen sclerosus, dermatomyositis, systemic and cutaneous lupus erythematosus), infections (viral, bacterial, fungal or parasitic) and neoplastic disorders (cutaneous malignancies).11
The treatment is, in general, insufficient and includes emollients, topical steroids, antihistamines, phototherapy and oral antihistamines. Definite resolution can only be achieved after treating the underlying disease.
Vesiculobullous Disorders
This group of disorders shows typical microscopic features namely epidermal cell separation, intraepidermal blister formation and deposition of immunoreactants in intercellular spaces of epithelial tissue.15 Its physiopathology is based on the production of autoantibodies that target specific proteins (desmogleins), responsible for the binding to the cell surface of keratinocytes of skin and mucous membranes. Pemphigus can be divided into two major subtypes: pemphigus vulgaris (the most common form), and paraneoplastic pemphigus.15 Microscopically, pemphigus vulgaris shows a pauci-inflammatory infiltrate with prominent acantholysis whereas paraneoplastic pemphigus has a marked mononuclear, lichenoid inflammatory infiltrate at the dermo-epidermal junction. Acantholysis is also present but in a lesser extent than in pemphigus vulgaris.15
In pemphigus vulgaris (PV) patients exhibit circulating IgG autoantibodies that target Desmoglein 3, located on the lower portion of epidermis, affecting primarily the mucous membranes throughout the body. Patients present with painful erosions of the mouth and mucous membranes of the pharynx, larynx, esophagus, eyes, nose and genitalia.16
Paraneoplastic pemphigus (PNP) has some features that allow us to distinguish it from PV, such as persistent and painful stomatitis, involvement of other mucosae and, in some cases, the presence of polymorphous, vesicular lichenoid skin eruptions (Fig 4). Also, in PNP the lesions typically involve the palms and soles whereas in PV this rarely occurs.17
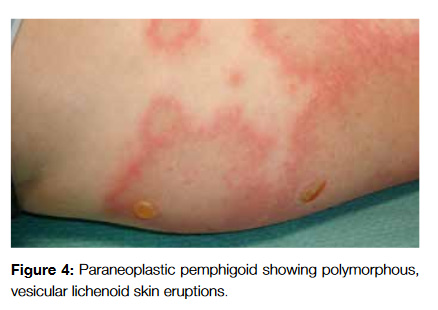
Anhalt et al proposed five classification criteria to define paraneoplastic pemphigus18:
1.Painful mucosal erosions and a polymorphous skin eruption with papular lesions progressing to blisters and erosive lesions affecting the trunk, extremities, palms and soles; in the context of an occult or confirmed neoplasia;
2.Cutaneous histologic changes such as intraepidermal acantholysis, keratinocyte necrosis and vacuolar-interface dermatitis;
3.Deposition of IgG and complement in the epidermal intercellular spaces, as well as granular-linear complement deposition along the epidermal basement membrane, on direct immunofluorescence testing;
4.Serum autoantibodies that bind the cell surface of skin and mucosae in a pattern typical of pemphigus but in addition bind to simple, columnar and transitional epithelia;
5.Presence of a complex of four proteins immunoprecipitated from keratinocytes by the circulating autoantibodies.
The most frequent malignancies in these patients are Kaposi sarcoma, Hodgkin and non-Hodgkin lymphoma, chronic lymphocytic leukemia,Waldenstrom macroglobulinemia and thymomas,15,17,19 being lymphoproliferative disease the most common one.15 The onset of pemphigus precedes the diagnosis of a malignancy in more than half of the cases.2
The differential diagnosis includes bullous pemphigoid, Hailey-Hailey disease (if there is localized oral disease), lichen planus, impetigo, subacute cutaneous lupus erythematosus and aphthous dermatitis.11
Regarding the treatment, most patients with PV have a good response to moderate or high-dose systemic corticosteroid therapy.15 Other therapies that can be used are intravenous immunoglobulin, plasmapheresis, corticosteroid-sparing drugs and rituximab.17 In paraneoplastic pemphigus, there is usually no response to any of these therapies, carrying a 90% mortality rate within a few months after diagnosis, independent of the course of the malignancy. In few reports, successful treatment of the associated neoplasia was associated with resolution on PNP.17
Amyloidosis
Amyloidosis is defined as in vivodeposition of material with specific characteristics: fibrillar appearance on electron microscopy; amorphous eosinophilic appearance on hematoxylin and eosin staining; beta-pleated sheet structure as observed by X-ray diffraction pattern; apple-green birefringence on Congo red staining, solubility in water and buffers of low ionic strength.20 All types of amyloid consist of one major fibrillar protein that defines the type of amyloid. Many mechanisms of protein function contribute to amyloidogenesis including defective or absent physiologic proteolysis, mutations involving changes in thermodynamic or kinetic properties and pathways that are yet to be defined.20
Primary amyloidosis is a type of amyloidosis in which no predisposing cause is found. It has a different distribution from its typical form, involving the skin and occurring in association with hematological malignancies. In contrast, secondary amyloidosis usually involves parenchymal structures and is associated with chronic inflammatory disorders.2
In terms of diagnosis, newer methods have been developed including subcutaneous fat biopsy. This is because recent studies have recognized the role of capillaries in the subcutaneous fat which are often involved in patients with systemic amyloidosis and can provide enough tissue for the diagnosis of amyloid, immunostaining, and, in some cases, amino acid sequence analysis.
Primary amyloidosis is due to a plasma cell dyscrasia that does not fulfill the criteria for multiple myeloma, being the precursor proteins immunoglobulin light chains rather than heavy chains. It has been associated with multiple myeloma (13-26%) and monoclonal gammopathy (90%).2 Patients with a multiple myeloma have a 16% probability of having amyloidosis due to deposition of light-chain elements.
Clinically, amyloidosis presents as firm skin-colored waxy papules or plaques occurring typically in the face; infiltration of the tongue can lead to macroglossia and skin trauma may cause the formation of purpura.11 Other manifestations are nephrotic syndrome, heart failure, splenomegaly, liver injury and hepatomegaly, peripheral neuropathy and mucocutaneous lesions.
The differential diagnosis of amyloidosis includes facial papules, mucinoses and multiple adnexal tumors.
The treatment is usually directed towards the main organ involved and the underlying etiology (chronic inflammatory disorder, malignancy), being the response very variable. The prognosis for myeloma-related amyloidosis is poor, with a survival of less than one year.2
Reactive Erythemas and Erythroderma
These are a group of cutaneous disorders characterized by migratory annular, arciform or polycyclic erythematous lesions, being erythema gyratum repens (EGR) the only entity considered to be a paraneoplastic disease. Similarly to erythroderma, they can occur in association with solid malignancies (namely EGR), such as lung, esophageal and breast cancer.21
Erythema gyratum repens (EGR), can be distinguished by rapidly evolving scaly, erythematous bands that form concentric rings resulting in a wood-grain appearance (Fig 5). They usually cover the trunk and proximal extremities and can be extremely pruritic. Their association with malignancy is well established, but there is not a predominance of any particular type of cancer (some of the reported cases were with lung cancer, Hodgkin lymphoma and multiple myeloma).21
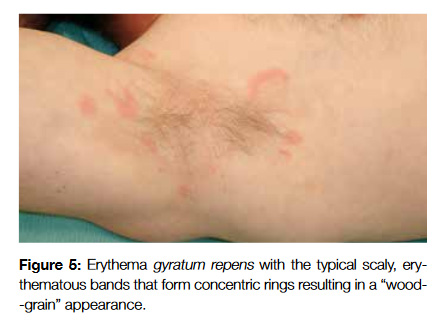
Erythroderma and exfoliative dermatitis typically present with generalized redness and scaling involving more than 90% of the body surface area. Lymphomas (Hodgkin and non-Hodgkin), leukemias and myelodysplastic syndromes are the most common malignancies linked to these disorders even though erythroderma is thought to be caused by the direct tumor invasion and not by a paraneoplastic process.2
The differential diagnosis of reactive erythemas includes drug eruptions, psoriasis vulgaris, pityriasis rubra pilaris, infections (HIV, Staphylococcus aureus), erythema chronicum migrans (usually accompanied by a history of a tick bite), annular urticaria, granuloma annulare, sarcoidosis, leprosy, secondary syphilis, tinea corporis, cutaneous T-cell lymphoma and subacute lupus erythematosus.21
The treatment is based on the identification of the underlying condition as the course of the cutaneous lesions follows the one of the underlying process. Systemic and topical corticosteroids provide symptom relief.
Vascular Dermatoses
The common feature of all the conditions is that they may occur as reactive processes in association with other diseases. The histopathologic characteristics of the lesions range from mild, perivascular dermal infiltration with inflammatory cells and vasodilation to various degrees of vessel damage (endothelial swelling to fibrinoid necrosis).22
Livedo reticularis is a common skin finding that consists of a mottled reticular blue-violet pattern reflecting an increase in deoxygenated blood within the venous plexus of the skin due to capillary obstruction by small clots. It can be due to arteriolar vasospasm or sluggish flow due to hypercoagulability or luminal pathology.11 It usually occurs on the lower limbs but, in the setting of systemic diseases, it may be widespread. It can be associated with several diseases, namely connective tissue diseases, Raynauds phenomenon, vasculitis, calciphylaxis, livedoid vasculopathy, hypercoagulability, polycythemia vera, thrombocythemia, infections, drugs or neoplasms (pheochromocytoma, multiple myeloma). It may improve with warming of the legs but in some cases, the discoloration may become permanent. The identification and treatment of the underlying disease is of crucial importance.
Erythromelalgia is a rare condition that occurs, in the majority of cases, in association with myeloproliferative diseases (59% in polycythemia vera, 38% in essential thrombocytosis).23 The diagnostic criteria comprise burning pain accompanied by increased cutaneous temperature and skin erythema of the lower extremities; precipitation of symptoms by heat, standing and exercising; relief of symptoms by cooling or elevation of the extremity.
It can be divided into early and adult-onset, according to the age of the patient, being the adult form further classified into primary or secondary. Early-onset erythromelalgia occurs in children, with a mean age of 10 years old and a female prevalence. It has a bilateral and symmetrical distribution and it commonly affects the ankles, feet and distal portion of lower legs. Adult-onset erythromelalgia can be idiopathic or primary if it is not associated with any other disease, having a male prevalence and a mean age of onset between 30 and 80 years old. There is also bilateral involvement of extremities and symptoms can be relieved by the use of aspirin. Secondary erythromelalgia is commonly associated with a myeloproliferative disorder (polycythemia vera, myelofibrosis, chronic myelogenous leukemia) and in 85% of patients; the cutaneous symptoms develop before the myeloproliferative disorder (average time of 2 and a half years). It has also been reported to occur in association with systemic lupus erythematosus, rheumatoid arthritis, pregnancy, diabetes mellitus, vasculitis and ingestion of certain drugs. It is thought to be caused by platelet hyperaggregability and activation that then leads to blockage of the arterioles.23
Vasculitis is a condition in which there is inflammation and fibrinoid necrosis of the blood vessel wall. There are several proposed classification systems, some focused on the vessel diameter and others based on histologic findings. It can also be further divided into primary or idiopathic and secondary (infections, chronic inflammation or neoplasms).
The systemic vasculitis can be classified according to the size of the vessels involved into small-vessel vasculitis (Wegeners granulomatosis, Churg-Strauss syndrome, microscopic polyarteritis, Henoch-Schonlein purpura, essential cryoglobulinemic vasculitis and cutaneous leukocytoclastic vasculitis), medium-sized vessel vasculitis (polyarteritis nodosa and Kawasaki disease) and large-vessel vasculitits (giant cell or temporal arteritis and Takayasu arteritis). All of them can have systemic symptoms, which include neurologic, renal, gastrointestinal, arthralgias and fever. The clinical and physiopathological features of each of them are very specific and extensive and beyond the purpose of this article.
The autoimmune etiology can be explained by three main mechanisms: immune complex deposition in vessel walls or in situ formation; direct autoantibody binding to antigens expressed in vessel walls and leukocyte activation by antibodies with specificity to certain antigens expressed in these cells (antineutrophil cytoplasmic autoantibodies).24 The most common vasculitis associated with malignancy are cutaneous leukocytoclastic vasculitis and polyarteritis nodosa, occuring in up to 5% of the patients.2
It has been postulated that tumoral cells can produce specific antigens that can interact with self-antigens and lead to the development of paraneoplastic vasculitis. The diagnostic criteria of paraneoplastic vasculitis proposed by McLeanet et al25 are the simultaneous appearance of both vasculitis and neoplasms and their parallel clinical course. Most of these occur in association with hematological malignancies, mostly lymphoproliferative diseases, hairy cell leukemia, Hodgkins disease, chronic myelogenous leukemia, myelodysplastic syndromes and multiple myeloma.2 Leukocytoclastic vasculitis is the most common one (30 to 40% of all paraneoplastic vasculitides). The direct or indirect action of malignant cells as sensitizing agents that release cytokines and damage the vascular endothelium is one of the proposed pathological mechanisms; however, further investigation needs to be done.25
The signs and symptoms of paraneoplastic vasculitis are similar to those in patients without an underlying malignancy and include unspecific cutaneous lesions (palpable purpura on the lower extremities, livedo reticularis, ulcers and digital finger necrosis), peripheral neuropathy, anasarca (secondary to nephrotic syndrome), asymmetric polyarthritis and fever.
The differential diagnosis includes recovery phase of frostbite, complex regional pain syndrome, autonomic dysfunction, hyper perfusion phase of Raynaud syndrome, peripheral vascular disease and cellulitis.23
The treatment of secondary erythromelalgia consists of a daily dose of 500 mg of acetylsalicylic acid. In the presence of vasculitis, corticosteroids alone or in association with imunossupressive agents are the drugs of choice.
Other Skin Disorders
These are cutaneous syndromes characterized by the presence of small patches, papules or large plaques with a raised border. Most of them are associated with solid tumors but acanthosis nigricans, sign of Leser-Trélat and acquired ichthyosis can be observed in hematological neoplasms.26-28
Acanthosis nigricans is defined by the rapid onset of hyper pigmented velvety changes of the flexural surfaces (neck, armpits, groin). It may also involve extensor surfaces (elbows, knees, knuckles) and, when it is associated with malignancy, the lips (in the form of glossitis), oral mucosa and palms are also involved.11 In approximately one third of cases of malignant acanthosis nigricans, patients present with skin changes before any signs of cancer. In another one third of cases, the lesions of acanthosis nigricans arise simultaneously with the neoplasm and in the remaining one third of cases, the skin findings manifest sometime after the diagnosis of cancer. Malignant acanthosis nigricans has been reported to appear abruptly and exuberantly and may be associated with a higher rate of pruritus.26
The sign of Leser-Trélat has been defined as the sudden appearance and rapid increase in size and number of seborrheic keratoses in association with the development of an internal malignancy.27 Initially assumed as an incomplete variant of acanthosis nigricans (already established as a marker of internal malignancy), it is nowadays thought to be due to a genetic abnormality which is only expressed in the presence of a malignant carcinoma. Additionally, the decreased host immunity and dysregulation on epidermal cell turnover are thought to contribute to the development of the cancer and of the seborrheic keratoses.27 The cutaneous lesions themselves are benign and do not require any specific treatment. Acquired ichthyosis (AI) is a condition characterized by dry, rough skin with prominent scaling, resembling fish scale. AI most commonly occurs in adulthood and manifests as symmetric scaling, with a coloration that can range from white to brown and primarily affecting trunk and extensor surfaces of the limbs. It occurs when there is a disruption in cornification, leading to hyperkeratosis, scaling and abnormalities on the barrier function of stratum corneum.28 AI has been described in association with Hodgkin and non-Hodgkin lymphomas, multiple myeloma, HIV infection, malnutrition and autoimmune disorders.28 The most commonly associated malignancy is Hodgkins disease, occurring in 70% to 80% of the cases.2 Once the diagnosis is established, the main purpose is the identification of the underlying disease. General treatment includes hydration, and keratolytics (salicylic acid, urea, lactic acid) and additional care is needed to prevent skin infections, as the barrier function is impaired.
Discussion
Cutaneous paraneoplastic syndromes may occur before, after or concomitantly with the cancer diagnosis. Their clinical features do not usually differ from the patients without a malignancy, being the patient history and careful clinical examination of essential importance.
The most commonly associated hematological malignancies are, in lymphoproliferative diseases, Hodgkin and non-Hodgkin lymphoma, chronic lymphocytic leukemia, multiple myeloma and Waldenstrom macroglobulinemia; in myeloproliferative disorders, acute myelogenous leukemia, myelodysplasic syndromes and primary myelofibrosis.
These syndromes usually show a favorable response to systemic corticosteroids but successful resolution typically occurs only after eradication of the underlying malignancy, exception being made to paraneoplastic pemphigus (which is refractory to therapy and usually shows an independent course from the malignancy). Overall, their presence is an indicator of a poor prognosis.17
Referencias
1.Carlesimo M, Narcisi A, Rossi A, Saredi I, Orsini D, Pelliccia S, et al. Cutaneous manifestations of systemic non-Hodgkin lymphomas (NHL): study and review of the literature. J Eur Acad Dermatol Venereol. 2014; 28:133-41. [ Links ]
2.Kurzrock R, Cohen PR. Mucocutaneous paraneoplastic manifestations of hematologic malignancies. Am J Med. 1995; 99:207-16. [ Links ]
3.Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweets syndrome. Postgrad Med J. 1997; 73:65-8. [ Links ]
4.Cohen PR, Kurzrock R. Sweets syndrome: a review of current treatment options. Am J Clin Dermatol. 2002; 3:117-31. [ Links ]
5.Cohen PR. Sweets syndrome – a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007; 2:34. [ Links ]
6.Luthra HS. Chapter 22: Relapsing Polychondritis. In: Klippel JH, Stone JH, Crofford LJ, White PH, editors. Primer on the rheumatic diseases. Atlanta: Springer,Arthritis foundation; 2007. p.451-3. [ Links ]
7.Myers B, Gould J, Dolan G. Relapsing polychondritis and myelodysplasia: a report of two cases and review of the current literature. Clin Lab Haematol. 2000; 22: 45-7. [ Links ]
8.András C, Ponyi A, Constantin T, Csiki Z, Szekanecz, Szodoray P, et al. Dermatomyositis and polymyositis associated with malignancy: a 21-year retrospective study. J Rheumatol. 2008; 35:438-44. [ Links ]
9.Marie I, Guillevin L, Menard J-F, Hatron PY, Cherin P, Amoura Z, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012; 11:615-20. [ Links ]
10.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet.2003; 362:971-82. [ Links ]
11.Bolognia JL, Schaffer JV, Duncan KO, Ko CJ. Dermatology Essentials. Philadelphia: Elsevier Saunders; 2014. [ Links ]
12.Yosipovitch G. Pruritus - an update. Curr Probl Dermatol. 2003; 15:135-64. [ Links ]
13.Yosipovitch G. Chronic pruritus: a paraneoplastic sign. Dermatol Ther.2010; 23: 590-6. [ Links ]
14.Krajnik M, Zylicz Z. Understanding pruritus in systemic disease. J Pain Symptom Manage. 2001; 21: 151-68. [ Links ]
15.Nguyen VT, Ndoye A, Bassler KD, Shultz LD, Shields MC, Ruben BS, et al. Classification, clinical manifestations and immunopathological mechanisms of the epithelial variant of paraneoplastic autoimmune multiorgan syndrome. Arch Dermatol. 2001; 137: 193-206. [ Links ]
16.Chaves MY, Pérez PU. Cutaneous manifestations of systemic malignancies: Part 2. Actas Dermosifiliogr. 2013; 104:543-53. [ Links ]
17.Anhalt GJ, Kim S, Stanley JR, Korman NJ, Jabs DA, Kory M, et al. Paraneoplastic pemphigus – an autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med. 1990; 323: 1729-35. [ Links ]
18.Steele HA, George BJ. Mucocutaneous paraneoplastic syndromes associated with hematologic malignancies. Oncology. 2011; 11: 1076-83. [ Links ]
19.Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999; 103: 461-8. [ Links ]
20.Medscape.com [homepage in the Internet] Amyloidosis; 2014 [assessed 23 February 2016]. Available from: http://emedicine.medscape.com/article/335414-overview#a1 [ Links ]
21.Tyring SK. Reactive erythemas: erythema annulare centrifugum and eythema gyratum repens. Clin Dermatol. 1993; 11:135-9. [ Links ]
22.Jorizzo JL. Classification of urticaria and reactive inflammatory vascular dermatoses. Dermatol Clin. 1985; 3:3-12. [ Links ]
23.Norton JV, Zager E, Grady JF. Erythromelalgia: Diagnosis and classification. J Foot Ankle Surg. 1999; 38:238-41. [ Links ]
24.Jenette CJ, Milling DM, Falk RJ. Vasculitis affecting the skin. Arch Dermatol. 1994; 130:899-906. [ Links ]
25.Sánchez NB, Canedo IF, García-Patos PE, Pérez PU, Pascual AM. Paraneoplastic vasculitis associated with multiple myeloma. J Eur Acad Dermatol Venereol. 2004; 18:731-5. [ Links ]
26.Sinha S, Schwartz RA. Juvenile acanthosis nigricans. J Am Acad Dermatol. 2007; 57:502-8. [ Links ]
27.Safal B, Grant JM, Good RA. Cutaneous manifestation of internal malignancies (II): sign of Leser-Trélat. Int J Dermatol. 1978; 17: 494-5. [ Links ]
28.Patel N, Spencer LA, English JC, Zirwas MJ. Acquired ichthyosis. J Am Acad Dermatol. 2006; 55:647-56. [ Links ]
Correspondência:Joana Azevedo Duarte joanaad88@gmail.com
Serviço de Medicina IV, Hospital Professor Doutor Fernando da Fonseca, Lisboa, Portugal
IC 19, 2720-276 - Amadora
Conflicts of interest: The authors have no conflicts of interest to declare.
Financing Support: This work has not received any contribution, grant or scholarship.
Confidentiality of data: The authors declare that they have followed the protocols of their work center on the publication of data from patients.
Protection of human and animal subjects: The authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Protecção de Seres Humanos e Animais: Os autores declaram que não foram realizadas experiências em seres humanos ou animais.
Direito à Privacidade e Consentimento Informado: Os autores declaram que nenhum dado que permita a identificação do doente aparece neste artigo.
Conflitos de Interesse: Os autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
Fontes de Financiamento: Não existiram fontes externas de financiamento para a realização deste artigo.
Recebido: 19/04/2016
Aceite: 21/07/2016

