










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.38 no.6 Coimbra set. 2020
https://doi.org/10.4152/pea.202006377
ARTIGOS
Evaluation of Anticorrosion Properties of 1, 2 ,4-triazole Derivatives on Steel in Acidic Media using Quantum Chemical Calculation and Molecular Dynamic Simulation Methods
U. Bello*, A. Uzairu and G. A. Shallangwa
Department of Chemistry, Ahmadu Bello University, Zaria, Nigeria
ABSTRACT
Molecular modelling approach has been used for the prediction of anticorrosion properties of 1, 2, 4-triazole derivatives on steel in acidic medium by the quantum chemical calculation and molecular dynamics (MD) simulation methods. Quantum chemical parameters such as the highest occupied molecular orbital (E-HOMO), the energy of the lowest unoccupied molecular orbital (E-LUMO), energy band gap (ΔE), dipole moment, global electronic chemical potential (μ), chemical softness (σ), chemical hardness (η) and electrophilicity (??) have been calculated and discussed. The reactive sites of the inhibitor molecules were found to be on the nitrogen-atom of the Triazolic ring and on the π-electron centers. Furthermore, molecular dynamics simulation was applied to search for the best inhibitor adsorption configuration over Fe (110) surface. The best adsorption energy was found to be -430.27 kcal/mol (inhibitor 3). The adsorptions occurred via chemisorption.
Keywords: molecular modelling, dynamic simulation, DFT, quantum chemical studies.
Introduction
Steel corrosion is a huge concern in oil industry, and results in an enormous waste of resources and potential safety issues. Nowadays, it is important to control metals corrosion, in order to prolong the life of the metallic equipment and to reduce the leaking of toxic metals to the environment.
Organic molecules with heteroatoms, mainly N, S, P or O and an aromatic ring, are commonly used as metal corrosion inhibitors (1, 2). Such organic inhibitors adhere onto the metallic surface by the physical or chemical adsorption route and, thereby, can prevent metallic corrosion.
Recently, tetrazole derivatives, triazole derivatives, thiadiazole derivatives, imidazole derivatives, cysteine and substituted uracils (3-8) were proved as excellent corrosion inhibitors in acidic media. Triazole and triazole-type molecules that contain sulfur and nitrogen have drawn more attention because of their excellent corrosion inhibition properties (9). New triazole derivatives have been repeatedly synthesized and investigated as inhibitors of metal corrosion in acidic solutions (10). The triazole derivatives have a unique affinity towards metal surfaces substituting water molecules on it. Additionally, they possess abundant π-electrons and unshared electron pairs on the nitrogen atom that can interact with d-orbitals of iron to provide a protective film.
Experimentally, the performance of inhibitive action has been assessed by weight loss measurements, potentiodynamic polarization and electrochemical impedance spectroscopy. However, these experimental methodologies are costly, time-consuming and sometimes deficient in explaining inhibition mechanisms at the sub-atomic and molecular levels (11).
In this occasion, appropriate molecular modeling and corresponding quantum chemical calculation are very efficient for exploring the relationship between the molecular properties of the inhibitors and their anti-corrosion properties (12). Corrosion inhibition ability of the molecules can be determined by the frontier molecular orbital energies, energy gap, dipole moment, global hardness, softness, etc. Previously, some researchers have successfully investigated the correlation between the quantum chemical calculations and experimentally obtained corrosion inhibition efficiency of the pyrazine derivatives, mercapto-quinoline derivatives and Schiff base molecules (13).
However, some researchers recently proposed that quantum chemical approach is not enough to envisage the inhibition efficiency trend of the inhibitors molecules (13). In many cases, results obtained from DFT cannot be well correlated with obtained experimental findings (14).
In this circumstances, a precise modeling of experimentation should be emphasized to correlate the theoretical results with the experimental inhibition effectiveness. In real practice, the modeling of an experiment can only provide the actual interfacial interactions between the concerned metallic surface and inhibitor molecules. As a result, recently molecular dynamics (MD) simulation has emerged as a modern tool from where we can reasonably predict actual interfacial configuration and adsorption energies of the surface adsorbed inhibitor molecules.
Until date, only a few certain groups have been working on this research to obtain the interaction, as well as the binding energy of surface adsorbed inhibitor molecules (1). Recently, some researchers have employed MD simulation to study the adsorption behavior of pyrazine derivatives onto the steel surface (15). Obot and Obi-Egbedi (2010) explored the correlation between the structural conformation of the imidazoline derivatives and their corresponding inhibition efficiencies by employing MD (16).
In the present investigation, we have successfully used both the quantum chemical calculation and MD simulations to explore the correlation between the theoretical results of 1, 2, 4-triazole derivatives and previously obtained experimental findings.
Materials and methods
Three 1,2,4-triazole derivatives studied by literature (17) were used as steel corrosion inhibitors in the present study. Their molecular structures and inhibition efficiencies are shown in table 1.
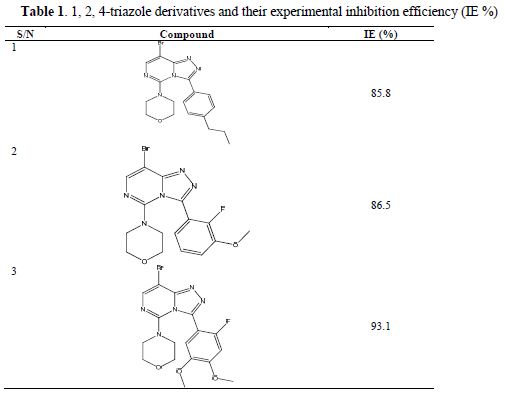
Molecular optimization and quantum chemical calculations
The chemical structure of each compound in the data sets was drawn with ChemDraw ultra V12.0. Optimization was performed using the SPARTAN’14 V1.1.0 WaveFunction programming package on Dell Intel(R)Core(TM)i7-5500U CPU), 16.00GB RAM @ 2.400GHz processor on Windows 8.1 Pro 64-bit operating system, ×64-based processor. The computational method invoked for calculating geometries in the present study is Density Functional Theory (DFT) method, in combination with the B3LYP functional of 6-311+G (d, p) basis set(18). The B3LYP hybrid functional of DFT method used Becke’s functional three-parameter (B3) and incorporated a blend of HF with DFT exchange terms associated with the gradient-corrected correlation functional of Lee, Yang, and Parr (LYP) (19). Quantum chemical parameters, such as energy of the highest occupied molecular orbital (E-HOMO), energy of the lowest unoccupied molecular orbital (E-LUMO), energy band gap (ΔE = E-LUMO – E-HOMO), dipole moment, global electronic chemical potential (μ), chemical softness (σ), chemical hardness (η) and electrophilicity (??) from the orbital energies (E-HOMO and E-LUMO), using appropriate relations (equations 1 to 5), as previously reported in literature (2), were computed.
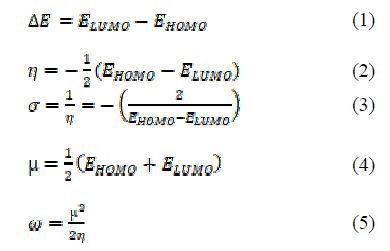
Molecular dynamics simulation computational method
The molecular dynamics simulation calculations were carried out to describe the interaction between the corrosion inhibitor molecules and the metallic surface using materials studio 8.0 software (Accelrys Inc., 2007). The inhibitors were modeled and optimized using the Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies (COMPASS) force field. COMPASS is a robust and well-developed force field that was derived based on fitting against a wide range of experimental data for organic and inorganic compounds (20). This informs its suitability for treating metal and non-metal containing systems. The whole system was performed at 298 K controlled by the Andersen thermostat with fixed number-volume-energy (microcanonical) ensemble, with a time step of 1.0 fs, simulation time of 50 ps, using the COMPASS force field. The MD simulation was carried out in a simulation box (24.82 Å ×24.82 Å ×45.27 Å) with periodic boundary conditions. The box includes a Fe slab, an acid solution layer and an inhibitor molecule. Fe (110) surface was selected as the studied surface, as it has a density packed surface and it was the most stable (21). The iron crystal contained ten layers, and seven layers near the bottom were frozen. The density of the acid solution layer was set at 1.0 g/cm3. The adsorption and binding energies in the solution were calculated by the following equation (22).
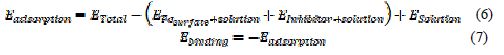
where ETotal was the total energy of the system, which includes iron crystal, the adsorbed inhibitor molecule and the solution; EFe_surface+solution and EFe_inhibitor+solution were the energies of the system without the inhibitor and the system without the iron crystal, respectively; and Esolution was the energy of the solution.
Results and discussions
Quantum chemical studies
Highest occupied molecular orbital (E-HOMO) and lowest unoccupied molecular orbital (E-LUMO) are very useful to elucidate the chemical reactivity of a molecule. According to the frontier molecular orbital theory of chemical reactivity, transition of electrons is mainly related to the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the reacting species (23). Fig. 1 shows the optimized structures, E-HOMOs and E-LUMOs of the studied inhibitors, respectively, and table 2 presents the calculated quantum chemical parameters.

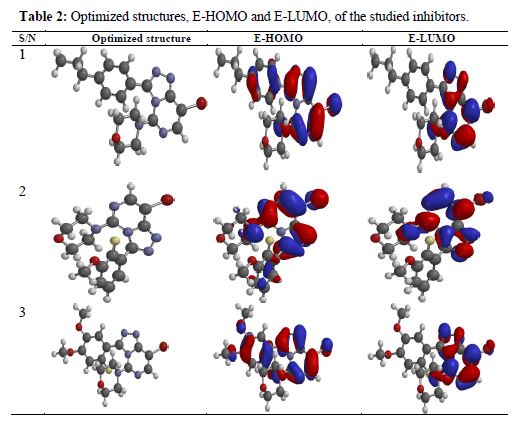
E-HOMO is associated to the electron donation capability of the molecule. Higher the E-HOMO value, stronger will be the electron donating capability of the inhibitor and, therefore, better the inhibition efficiency will be (12). LUMO implies the capability of the molecules to accept electrons from the metallic surface. Lower the value of E-LUMO, more it will be prone to accept electrons (1). From table 2, it can be seen that E-HOMO value increased in the order of 1< 2 < 3, which means that the ability to donate electrons from the inhibitor molecule to the metallic surface obeys the order 3 > 2 >1. The calculated E-LUMO value exemplifies that the electron acceptance capability decreased in the order of 3 > 2 > 1. Hence, the orders (E-HOMO and E-LUMO) are strongly correlated with experimental inhibition efficiencies.
Apart from E-HOMO and E-LUMO, energy gap (ΔE) is also an important parameter in determining the adsorption of organic molecules onto the metallic surface. As ΔE decreases, the reactivity of the molecule will definitely increase, which, in turn, leads to an increase in the adsorption onto a metallic surface (24). In general, molecules with comparatively lower energy gap are better polarizable and, in turn, are associated with higher chemical reactivity and lower kinetic stability. In this connection, ΔE has been used to elucidate the binding ability of the inhibitor molecule onto the metallic surface. It can be seen (table 2) that the ΔE value follows the trend 1 >2 > 3, which is again also well in accordance with the result obtained from the experimental outcomes.
The dipole moment of a molecule is also an important parameter to elucidate the chemical reactivity of a molecule. A literature survey reveals that the adsorption process is more facilitated with increasing values of the dipole moment, as it is influences the transport process through the adsorbed layer (23, 25). It can be observed from table 3 that the dipole moment values increased in the order of 1 < 2 < 3, which further more strengthens the experimental results. Global (chemical) hardness (η) and softness (σ) have all been used as molecular descriptors of reactivity and selectivity (26). Adsorption usually occurs in the region of the molecule where σ has the highest value. The order across structures in the σ values, as reported in table 3, suggests that the inhibitor-3 is the most reactive compound, followed by 2, then by 1, which agrees with the experimental findings.
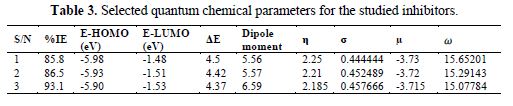
Global electronic chemical potential (μ) is often reported as electronegativity (χ) in most previous studies (23). According to them, electronegativity is often described as the negative global electronic chemical potential. The values of μ reported in table 3 show that their order is 3 >2 >1. This implies that 3 are the greater electron acceptor, which agrees with their high experimental inhibition efficiency. Electrophilicity index (ω) is another global reactivity index that is often used for the prediction of the direction of a corrosion inhibition process (13). A high electrophilicity value describes a good electrophile, while a small electrophilicity value describes a good nucleophile. The order across structures in the ω values, as reported in table 3, shows that the inhibitor-3 has the lowest and the highest experimental inhibition efficiency.
Molecular dynamics simulation
Electronic properties alone are not sufficient to predict the trend of the inhibition performance of the investigated inhibitors, although they help to explore the mechanism of inhibitors. Therefore, it is imperative to carry out rigorous modelling of the direct interaction of the inhibitors with Fe. It is generally believed that the primary mechanism of corrosion inhibition is adsorption. The adsorption of the studied inhibitors onto the steel surface was simulated by modeling the interactions between the inhibitors and Fe (110) crystal surface in 1 M HCl. The obtained E-adsorption and E-binding values, calculated according to equation (6) and (7), are presented in table 4. The adsorption configurations of the inhibitor molecules over the Fe (1 1 0) surface are shown in Fig. 1. It can be seen from this figure that inhibitor molecules adsorb almost on a flat orientation with respect to the iron surface. This flat orientation can be explained in terms of chemical bond formation between the inhibitors and the iron surface.
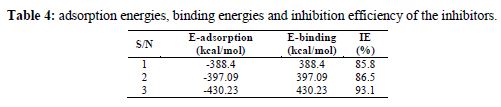
Generally, a chemical adsorption will occur on Fe surfaces if the binding energy is >100kcal/mol (27). Thus, it is confirmed from the MD simulations that the adsorption of the inhibitor molecules onto the metallic surfaces mainly occurred by the chemical adsorption phenomenon. Additionally, it could be seen in table 4 that the calculated adsorption energy values of the interaction systems at 298 K are -388.4, -397.09 and -430.23kcal/mol, respectively. These larger negative values of adsorption energies can be ascribed to the strong interaction between the studied inhibitors molecules and iron surfaces. Thus, calculated adsorption energy values reveal that inhibitor-3 molecules adsorb onto the iron surface more spontaneously than those of 2 and 1. Moreover, the adsorption ability of the molecules onto the iron surface can also be measured from the binding energy values. Higher is the binding energy, more strong will be the adsorption. Thus, it can be seen from the adsorption energy, as well as from binding energy values, that the adsorption ability of the inhibitors molecules onto the iron surface at 298 K follows the order 3 > 2 > 1. These outcomes are in good agreement with the results obtained from wet chemical experimentation.
Thus, in conclusion, it can be said that these results are in good agreement with the results obtained from wet chemical experimentation, as well as from quantum chemical calculations.
Conclusion
Molecular modelling theoretical analysis was carried out to study the anticorrosion properties of three 1, 2, 4- triazole derivatives onto a steel surface. It is evident from this investigation that only theoretical studies can provide a complete insightfulness into the chemical reactivity of the studied inhibitor molecule. Quantum chemical calculation reveals that electron donation and electron acceptance capability of the studied inhibitors follows the order 3 > 2 > 1, which is in good accordance with the results obtained from previously performed experimental findings.
MD simulation reveals that the adsorption occurred via chemical adsorption. An adsorption energy and binding energy value of the three studied inhibitors also obeys the order of 3 > 2 > 1. These outcomes are in good accordance with the experimental findings. The above mentioned results obtained from two different domains starting from density functional theory (based on quantum chemistry) to MD simulation (based on classical physics) are in fine agreement with the previously obtained experimental results. It can be concluded that DFT and MD simulations may be very powerful tools for the rational designing of several promising corrosion inhibitors and prediction of their inhibition efficiency well in advance.
References
- Wazzan NA, Obot I, Kaya S. Theoretical modeling and molecular level insights into the corrosion inhibition activity of 2-amino-1, 3, 4-thiadiazole and its 5-alkyl derivatives. J Mol Liq. 2016;221:579.
- Nwankwo HU, Olasunkanmi LO, Ebenso EE. Experimental, quantum chemical and molecular dynamic simulations studies on the corrosion inhibition of mild steel by some carbazole derivatives. Sci Rep. 2017;7(1):2436.
- Collins W, Weyers R, Al-Qadi I. Chemical treatment of corroding steel reinforcement after removal of chloride-contaminated concrete. Corrosion. 1993;49(1):74.
- Dafali A, Hammouti B, Mokhlisse R et al. Substituted uracils as corrosion inhibitors for copper in 3% NaCl solution. Corros Sci. 2003;45(8):1619.
- Es-Salah K, Keddam M, Rahmouni K et al. Aminotriazole as corrosion inhibitor of Cu-30Ni alloy in 3% NACl in presence of ammoniac. Electrochim Acta. 2004;49(17-18):2771.
- Finšgar M, Milošev I. Inhibition of copper corrosion by 1, 2, 3-benzotriazole: a review. Corros Sci. 2010;52(9):2737.
- Ferreira E, Giacomelli C, Giacomelli F, et al. Evaluation of the inhibitor effect of L-ascorbic acid on the corrosion of mild steel. Mater Chem Phys. 2004;83(1):129.
- Ezeoke AU, Adeyemi OG, Akerele OA, et al. Computational and experimental studies of 4-Aminoantipyrine as corrosion inhibitor for mild steel in sulphuric acid solution. Int J Electrochem Sci. 2012;7:534.
- Lebrini M, Robert F, Roos C. Alkaloids extract from Palicourea guianensis plant as corrosion inhibitor for C38 steel in 1 M hydrochloric acid medium. Int J Electrochem Sci. 2011;6(3):847.
- Martinez S, Metikoš-Hukovic M. A nonlinear kinetic model introduced for the corrosion inhibitive properties of some organic inhibitors. J Appl Electrochem. 2003;33(12):1137.
- Obot I, Macdonald D, Gasem Z. Density functional theory (DFT) as a powerful tool for designing new organic corrosion inhibitors. Part 1: an overview. Corros Sci. 2015;99:1.
- Saha SK, Banerjee P. A theoretical approach to understand the inhibition mechanism of steel corrosion with two aminobenzonitrile inhibitors. RSC Adv. 2015;5(87):71120.
- Wazzan NA. DFT calculations of thiosemicarbazide, arylisothiocynates, and 1-aryl-2, 5-dithiohydrazodicarbonamides as corrosion inhibitors of copper in an aqueous chloride solution. J Ind Eng Chem. 2015;26:291.
- Verma C, Olasunkanmi LO, Ebenso EE, et al. Adsorption behavior of glucosamine-based, pyrimidine-fused heterocycles as green corrosion inhibitors for mild steel: experimental and theoretical studies. J Phys Chem C. 2016;120(21):11598.
- Singh P, Ebenso EE, Olasunkanmi LO, et al. Electrochemical, theoretical, and surface morphological studies of corrosion inhibition effect of green naphthyridine derivatives on mild steel in hydrochloric acid. J Phys Chem C. 2016;120(6):3408.
- Obot I, Obi-Egbedi N. An interesting and efficient green corrosion inhibitor for aluminium from extracts of Chlomolaena odorata L. in acidic solution. J Appl Electrochem. 2010;40(11):1977.
- Gurudatt DM, Mohana KN, Tandon HC. Synthesis of new 1, 2, 4-triazole derivatives and their anticorrosion properties on mild steel in hydrochloric acid medium. J Mol Liq. 2015;211:275.
- Larif M, Chtita S, Adad A et al. Predicting biological activity of Anticancer Molecules 3-ary l-4-hydroxyquinolin-2-(1H)-one by DFT-QSAR models. International J. 2013;3(12):32.
- Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98(7):5648.
- Wymyslowski A, Iwamoto N, Yuen MMF, et al. Molecular Modeling and Multiscaling Issues for Electronic Material Applications: Volume 2. 2014.
- Khaled K. Molecular simulation, quantum chemical calculations and electrochemical studies for inhibition of mild steel by triazoles. Electrochim Acta. 2008;53(9):3484.
- Zhao H, Zhang X, Ji L et al. Quantitative structure–activity relationship model for amino acids as corrosion inhibitors based on the support vector machine and molecular design. Corros Sci. 2014;83:261.
- Murulana LC, Singh AK, Shukla SK, et al. Experimental and quantum chemical studies of some bis (trifluoromethyl-sulfonyl) imide imidazolium-based ionic liquids as corrosion inhibitors for mild steel in hydrochloric acid solution. Ind Eng Chem Res. 2012;51(40):13282.
- Ebenso EE, Khaled K, Shukla SK, et al. Quantum chemical investigations on quinoline derivatives as effective corrosion inhibitors for mild steel in acidic medium. Int J Electrochem Sci. 2012;7:5643.
- Tanak H, Agar A, Yavuz M. Experimental and quantum chemical calculational studies on 2-[(4-Fluorophenylimino) methyl]-3, 5-dimethoxyphenol. J Mol Model. 2010;16(3):577.
- Abdallah M, Atwa S, Salem M, et al. Synergistic effect of some halide ions on the inhibition of zinc corrosion in hydrochloric acid by tetrahydro carbazole derivatives compounds. Int J Electrochem Sci. 2013;8:10001.
- Akalezi CO, Enenebaku CK, Oguzie EE. Application of aqueous extracts of coffee senna for control of mild steel corrosion in acidic environments. Int J Ind Chem. 2012;3(1):13.
Received June 7, 2018; accepted August 10, 2020
* Corresponding author. E-mail address: abdallahbum@yahoo.com
Acknowledgements
Department of Chemistry, Ahmadu Bello University, Zaria is gratefully acknowledged for getting the computational softwares for performing the DFT and MD calculations. The authors received no funds for this research.

