









Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortugaliae Electrochimica Acta
Print version ISSN 0872-1904
Port. Electrochim. Acta vol.37 no.6 Coimbra Dec. 2019
https://doi.org/10.4152/pea.201906345
ARTIGOS
Electroanalytical and Spectrophotometric Determination of N-acetyl-p-aminophenol in Pharmaceuticals
E. Goclik, E. Chrzescijanska, E. Kusmierek and J. Rynkowski
Lodz University of Technology, Faculty of Chemistry, Institute of General and Ecological Chemistry, 90-924 Lodz, ul. Zeromskiego 116, Poland
ABSTRACT
Electrochemical oxidation of N-acetyl-p-aminophenol (AAP) and selected pharmaceutical products containing AAP is described in this study. Investigations were carried out at Pt electrode, with the application of cyclic (CV) and differential pulse (DPV) voltammetry. AAP was irreversibly oxidized in, at least, one electrode step, with potentials lower than the potential at which oxygen evolution started. Electrochemical methods based on voltammetric techniques (DPV and CV) have been developed for AAP determination in commercial pharmaceutical drugs. AAP content in pharmaceuticals was determined on the base of dependences of current intensity (electroanalytical measurements) and absorbance (UV-VIS spectra). The obtained results using both methods were in good agreement. The validation of CV, DPV and spectrophotometric methods is also presented. Spectrophotometric and voltammetric responses linearly increased with increasing AAP concentrations, in the range from 0.2 to 5 mmol L-1, with a correlation coefficient of 0.997 (UV-VIS), 0.999 (CV) and 0.998 (DPV). Both electrochemical methods are simple, reliable and sufficiently accurate and precise for AAP quantification in commercial drug samples (tablets). The analysis took only 5 minutes of manual operation, including Pt electrode pre-treatment.
Keywords: Cyclic voltammetry (CV); differential pulse voltammetry (DPV); N-acetyl-p-aminophenol; pharmaceuticals products; UV-VIS spectrophotometry.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) belong to a non-uniform (taking into consideration the chemical structure) group of drugs with analgesic, antipyretic and anti-inflammatory activities. NSAIDs have already been used in ancient ages, in the form of extracts from a willow [1]. Since the introduction of the first synthesized NSAID - salicylic acid (SA) -, still used in the pharmaceutical industry as a component of drugs and cosmetics [2-5] -, many new anti-inflammatory drugs with higher therapeutic activity have been developed [6]. Among NSAIDs, available without prescription, the drug known as paracetamol is probably the most often applied. It contains an active substance, N-acetyl-p-aminophenol, with the IUPAC name N-(4-hydroxyphenyl)acetamide) (AAP), [7-8], of which average content in pharmaceuticals is usually at the level of 500 mg [9]. Pharmaceuticals containing AAP are widely used, because they are very effective in the treatment of colds, flu and pain [10]. Although paracetamol is generally regarded as safe for use, one should bear in mind that its application, especially over dosage, can lead to numerous undesired side effects: among others, accumulation of toxic metabolites resulting in hepatotoxicity [11-14] and nephrotoxicity [15], as well as cryptorchidism and bronchial asthma in developing embryos [16-17]. Taking into consideration the common usage of pharmaceuticals containing AAP, quick and sensible analytical methods should be developed in order to determine its content in drugs and body fluids, as well as to estimate the effects of NSAIDs on human body and health.
Many chromatographic methods, including gaseous and liquid chromatography, coupled with mass spectrometry (GC-MS, LC-MS) [18-21] and high-performance liquid chromatography (HPLC) [22-23], have been applied for AAP determination in pharmaceuticals. These methods allow qualitative and quantitative analysis of a tested substance. However, they require preliminary sample preparation, e.g., by application of various extraction methods and derivatization, which make their analysis time-consuming and complicated [23-25]. Spectrophotometric and spectrofluorometric methods are also often used in NSAIDs quantitative determination. These methods are characterized by their simplicity of analysis, and are less expensive and time-consuming, in comparison to other analytical methods [26].
Recently, Behera et al. [27] developed a spectrophotometric method of AAP content determination in pharmaceutical tablets. In the last years, electrochemical methods have drawn attention [28-31], due to their high sensitivity and accuracy, which make them convenient for the qualitative and quantitative analysis of organic compounds contained in pharmaceuticals [32-36]. They are characterized by simple measurements and short times of sample analysis in low cost apparatus, in comparison to chromatographic methods [37-40]. The most common electrochemical methods include differential pulse (DPV) and cyclic (CV) voltammetry [41-43]. DPV is characterized by higher resolution than CV, which enables better separation of peaks attributed to subsequent steps of electrode reactions. Both electroanalytical methods make possible the determination of the peak potential (Ep) and half-wave potential (E1/2). These parameters are especially important, due to the fact that their lower values indicate the higher ability of a tested compound to capture free radicals. This means better anti-oxidative properties of the tested compound [34, 44-47].
The aim of this investigation was to develop a simple and accurate electrochemical method for the determination of the active substance containing AAP in the tested pharmaceuticals, which are named apap, panadol and paracetamol – and are available in pharmacists and chemist’s shops –, using differential pulse (DPV) and cyclic (CV) voltammetry. AAP exhibits an irreversible oxidation peak at Pt electrode, which can be used for its quantitative determination. The practical application of the electroanalytical methods was demonstrated by determining AAP concentrations in different commercial tablets. The methods were validated taking into consideration the following parameters: limit of detection (LOD), limit of quantification (LOQ), precision and accuracy. The suggested methods are characterized by the no time-consuming sample preparation and Pt electrode pre-treatment steps prior to AAP determination.
Experimental
Reagents
The subject of the investigation was an active substance: N-acetyl-p-aminophenol (AAP, 99.0% purity), purchased at Sigma-Aldrich (Germany). Pharmaceutical products (apap, panadol, paracetamol) containing the tested compound were purchased at pharmacies in Lodz (Poland). According to the producers, each of the tested pharmaceuticals contained 500 mg of the active substance, and other auxiliary substances.
The AAP solutions concentrations varied in the range from 0.2 to 5.0 mmol L-1. The AAP aqueous solutions were prepared by their dissolution in 0.1 mol L-1 NaClO4 (Fluka, France). Pharmaceutical products (one tablet) were grounded to powder, and then dissolved in NaClO4.
Measurement methods
Voltammetric analysis
The electrochemical behaviour of the active substance (AAP) in pharmaceuticals was studied with the application of an Autolab PGSTA30 Electrochemical Analyzer (EcoChemie, The Netherlands). The analyser was controlled using GPES program. A three-electrode electrochemical cell employed in measurements consisted of a reference electrode, an auxiliary electrode (platinum wire) and a working electrode – platinum –, with a geometric surface area of 0.5 cm2. The potential of the working electrode was measured vs. the saturated calomel electrode (SCE). Before each measurement, the Pt electrode was pre-treated by thoroughly rinsing it in deionised water, followed by electrochemical treatment in H2SO4 (0.5 mol L-1). Ten cycles were recorded in the potential range from -0.25 to 1.0 V (vs. SCE), with the scan rate of 0.1 V s-1. Whenever an unchanged CV characteristic of the Pt electrode was observed, the electrode was removed, rinsed with deionised water and placed in the AAP solution. Determination of AAP behaviour was performed using cyclic (CV) and differential pulse (DPV) voltammetry. Cyclic voltammograms were recorded in the potential range from 0 to 1.0 or 1.4 V, with the scan rate of 0.01 V s-1. Differential pulse voltammograms were recorded in the same potential range, with a modulation amplitude of 25 mV and pulse width of 50 ms (scan rate of 0.01 V s-1). Before the measurements, the solutions were purged with argon, in order to remove dissolved oxygen. During measurements, an argon blanket was kept over the solutions. All experiments were carried out at room temperature.
Spectrophotometric analysis
UV-VIS spectra were recorded in AAP and pharmaceutical solutions, in the wavelength range from 190 to 800 nm, with the application of an UV 24001PC (Shimadzu, Japan) spectrophotometer. Before measurements, all analysed samples were 50 times diluted using 0.1 mol L-1 NaClO4.
Results and discussion
Electrochemical oxidation of AAP
The electrochemical oxidation behaviour of AAP was studied at a platinum electrode, with the application of CV and DPV methods. Exemplary voltammograms are presented in Fig. 1. Voltammograms were recorded in the potential range from 0 to 1.0 V, in which the supporting electrolyte (0.1 mol L-1 NaClO4) showed no peaks (Fig. 1, curve 3). Voltammograms presented in Fig. 1 (curves 1 and 2) show that AAP was probably irreversibly oxidized in, at least, one electrode step, at potentials lower than the potential at which oxygen evolution started. As it was previously stated [48], this step had a diffusive character. The half-wave potential (E1/2) of AAP electro-oxidation determined from the cyclic voltammogram (CV) had a total of 0.59 V, and corresponded to the peak potential (Epa) determined from the differential pulse voltammogram (DPV).
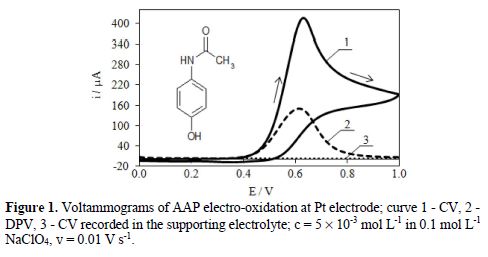
Kinetic parameters of this step, as well as an effect of different parameters (among others, scan rate, substrate concentration and pH) on the electrode reaction, were previously determined and described [48].
Electrochemical behaviour of the tested substance in pharmaceutical products
CV and DPV curves recorded in solutions of apap, panadol and paracetamol are presented in Fig. 2.
Voltammograms recorded for apap, panadol and paracetamol solutions show that these compounds were irreversibly oxidized in, at least, one electrode step, at potentials lower than the potential at which oxygen evolution started (Fig. 2A and 2B). The half-wave potential (E1/2) determined on the base of cyclic voltammograms had totals of 0.632 V (apap), 0.639 V (panadol) and 0.635 V (paracetamol). This means that apap was slightly easier oxidized than paracetamol and panadol. E1/2 is an especially important parameter, due to the fact that its lower value indicates the higher ability of a tested compound to capture free radicals, which means it has better anti-oxidative properties. The anti-oxidative properties of the tested pharmaceuticals increase in the following order: panadol < paracetamol < apap.
The dependences of the peak currents vs. AAP concentrations (Fig. 3) were linear, and are described by the following equations:
ip = {(79.607±7.9)(c(mM))} µA – (2.170±0.2) µA, R2 = 0.999 CV (1)
ip = {(28.295±2.8)(c(mM))} µA + (6.784±0.7) µA, R2 = 0.998 DPV (2)
These equations could be applied for the determination of AAP contents in the tested pharmaceuticals. The results of the content calculations of AAP are presented in Table 1.
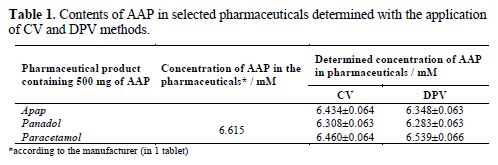
The concentrations of the active substance – in the tested pharmaceuticals – (determined with CV and DPV methods) were similar, but they were slightly lower than the values provided by the manufacturer. This can be explained by the effect of the auxiliary substances. This effect was the highest, in the case of panadol, and the lowest, in the case of paracetamol (Table 1).
UV-VIS spectrophotometric analysis of AAP and pharmaceutical products
AAP determination in the pharmaceuticals was also carried out with the application of the spectrophotometric method, similarly to how it was performed in the case of salicylic and acetylsalicylic acids [49-50].
UV-VIS spectra were recorded in AAP solutions, as well as in pharmaceuticals (apap, panadol, paracetamol), in the wavelength range from 180 to 800 nm, and are presented in Fig. 4. Fig. 4A shows three characteristic absorption bands at 205, 243 and 290 nm, in the AAP solution, with various concentrations. The absorption band at 243 nm (II band) is attributed to C=C bonds in the aromatic ring (π-π* transition). The first (205 nm) and third band (290 nm) resulted from interactions between the substituents in the AAP molecule and the aromatic ring [51-52]. Thus, a calibration curve presenting a dependence of absorbance on AAP concentration was determined for the absorption band at 243 nm. This dependence was linear (Fig. 4C) and could be successfully applied for the determination of AAP concentrations. Similar absorption bands were recorded in the solutions of the tested pharmaceuticals (Fig. 4B). The concentrations of the active substance in the pharmaceuticals calculated on the base of the calibration curve are presented in Table 2. Similarly, as in the case of voltammetric determinations, AAP concentrations determined with the spectrophotometric method are close to the values provided by the manufacturer. The highest difference in the concentration was observed in the case of panadol.
Method validation in AAP quantitative determination
AAP determination in pharmaceuticals, carried out with CV, DPV and spectrophotometric methods, was validated taking into consideration the following parameters: limit of detection (LOD), limit of quantification (LOQ), precision and accuracy. CV and DPV methods could be applied for AAP determination, because well-defined anodic peaks could be observed. The plots of peak currents versus AAP concentrations were found to be linear in the tested concentrations range. In the case of the spectrophotomeric method, the absorption band with the maximum at 243 nm could also be used for AAP determination.
LOD and LOQ
LOD is the lowest concentration (amount) of an analyte in a sample, e.g., pharmaceutical, which can be reliably distinguished from the noise level. LOQ is the lowest concentration of an analyte at which it can be reliably detected at a specified level of precision or accuracy (or both). These parameters can be calculated according to the following equations [27, 53-57]:
where SD is the standard deviation of replicate determination values under the same conditions as for the sample analysis in the analyte absence, and a is the slope of the calibration curve.
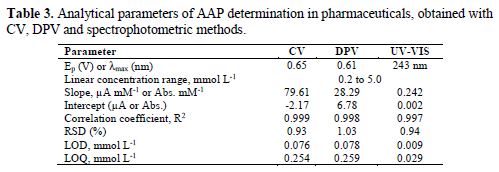
Results of the validation of the three applied determination methods are presented in Table 3. The values of LOD and LOQ are comparable for both voltammetric (CV and DPV) methods applied in AAP determination. The precision of the tested methods was determined by analysing 10 replicate measurements of the sample with the specified AAP concentration. The percentage of the relative standard deviation (RSD) values was about 1% for all applied methods.
Accuracy
The accuracy of an analytical method expresses the closeness of agreement between the value which is accepted either as a conventional true value or an accepted reference value and the found value [55-58]. The accuracy of the tested methods was estimated taking into consideration the percentage relative error (Err) calculated according to the following equation:
where xi is the determined concentration of the active substance in the pharmaceuticals and x0 is the nominal concentration of the active substance provided by the manufacturer. The results of Err calculations are presented in Table 4.
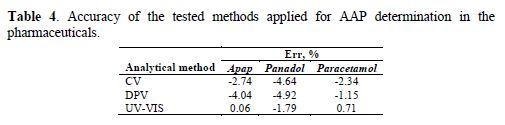
Results presented in Table 4 prove that the percentage relative error (Err) is the lowest in the case of paracetamol determination with the application of all tested methods. In the case of apap and panadol determination, the value of Err did not exceed 5%.
Conclusions
Electro-oxidation of pure N-acetyl-p-aminophenol (AAP) and selected pharmaceuticals (apap, panadol and paracetamol) containing AAP irreversibly proceeded at Pt electrode and in, at least, one electrode step, at potentials lower than the potential at which oxygen evolution started. A comparison of E1/2 values proved that apap revealed the highest ability to capture free radicals and the best anti-oxidative properties. Paracetamol and panadol showed slightly worse anti-oxidative properties.
The electrochemical methods based on voltammetric techniques (DPV and CV) have been successfully developed for AAP determination in pharmaceutical formulations (tablets). The results of electroanalytical determinations have been compared to results obtained by spectrophotometric analysis. The practical application of the tested methods was demonstrated by determining the concentration of AAP in commercial drugs. Dependences of peak currents and absorbances at 243 nm on AAP concentrations were linear in the tested concentrations range. AAP concentrations in the tested pharmaceuticals were found to be slightly different in comparison with data provided by the manufacturers. The validation showed acceptable linearity, precision, accuracy, reproducibility and sensitivity of all the three methods. The relative standard deviation (RSD) was about 1%. The accuracy determined for all methods was the lowest (Err < 2.5%) in the case of paracetamol, while Err did not exceed 5% in the case of apap and panadol. The UV-VIS spectrophotometry appeared to be the most accurate method for the determination of the tested pharmaceuticals. The main advantage of DPV and CV techniques over the other techniques is that they may be directly applied to the analysis of pharmaceuticals, without the need of separation or complex samples preparation. This results from the fact that no interference from excipients and the active substance was observed. The elaborated methods are rapid, with a sample running of about 5 min. Electrochemical methods seemed to be of particular importance, because they enabled not only quantitative determinations, but also got to reveal the electrochemical behaviour of the organic compounds, e.g., their anti-oxidative properties.
Thus, the tested methods could be helpful in pharmacokinetic investigations and therapeutic drug monitoring in body fluids.
References
- Sostres C, Gargallo CJ, Lanas A. Nonsteroidal anti-inflammatory drugs and upper and lower gastrointestinal mucosal damage. Arthritis Res Ther. 2013;15:S3.
- Chou WL, Wang ChT, Huang KF, et al. Electrochemical removal of salicylic acid from aqueous solutions using aluminum electrodes. Desalination 2011;271:55–61.
- Campanella L, Gregori E, Tomassetti M. Salicylic acid determination in cow urine and drugs using a bienzymatic sensor. J Pharmaceut Biomed Anal. 2006;42:94–99.
- Andreozzi R, Raffaele M, Nicklas P. Pharmaceuticals in STP effluents and their solar photodegradation in aquatic environment. Chemosphere. 2003;50:1319–1330.
- Ehrendorfer M, Sontag G, Pittner F. Determination of salicylate in beverages and cosmetics by use of an amperometric biosensor. Fresenius J Anal Chem. 1996;356:75–79.
- Miedzybrodzki R. Kierunki poszukiwan i zastosowanie niesteroidowych leków przeciwzapalnych. Postepy Hig Med Dosw. 2004;58:438–448.
- Li Y, Chen SM. The Electrochemical Properties of Acetaminophen on Bare Glassy Carbon Electrode. Int J Electrochem Sci. 2012;7:2175–2187.
- Daneshegar P, Moosavi-Movahedi AA, Norouzi P, et al. Characterization of Paracetamol Binding with Normal and Glycated Human Serum Albumin Assayed by a New Electrochemical Method. J Braz Chem Soc. 2012;23:315–321.
- Kujawska M, Murias M. Paracetamol–etanol – zlozona interakcja. Adv Clin Exp Med. 2004;13:497–501.
- Wang SF, Xie F, Hu RF. Carbon-coated nickel magnetic nanoparticles modified electrodes as a sensor for determination of acetaminophen. Sens Actuators B: Chem. 2007;123:495–500.
- Olaleye MT, Rocha BTJ. Acetaminophen-induced liver damage in mice: Effects of some medicinal plants on the oxidative defense system. Exp Toxicol Pathol. 2008;59:319–327.
- Roberts JC, Phaneuf HL, Szakacs JG, et al. Differential Chemoprotection against Acetaminophen-Induced Hepatotoxicity by Latentiated L-Cysteines. Chem Res Toxicol. 1998;11:1274–1282.
- Yayla M, Halici Z, Unal B, et al. Protective effect of Et-1 receptor antagonist bosentan on paracetamol induced acute liver toxicity in rats. Eur J Pharmacol. 2014;726:87–95.
- Chun LJ, Tong MJ, Busuttil RW, et al. Acetaminophen Hepatotoxicity and Acute Liver Fail. J Clin Gastroenterol. 43;2009:342–349.
- Mazer M, Perrone J. Acetaminophen-Induced Nephrotoxicity: Pathophysiology, Clinical Manifestations, and Mana. J Med Toxicol. 2008;4:2–6.
- Stosic R, Dunagan F, Palmer H, et al. Responsible self-medication: perceived risks and benefits of over-the-counter analgesic use. Int J Pharm Pract. 2011;19:236–245.
- Mrówka-Kata K, Czecior E, Kata D, et al. Current view on nasal polyps management in Samter’s triad Patients. Otolaryngol Pol. 2012;66:373–378.
- Weigel S, Kallenborn R, Hühnerfuss H. Simultaneous solid-phase extraction of acidic, neutral and basic pharmaceuticals from aqueous samples at ambient (neutral) pH and their determination by gas chromatography–mass spectrom. J Chromatogr A. 2004;1023:183–185.
- Gros M, Petrovic M, Barceló D. Multi-residue analytical methods using LC-tandem MS for the determination of pharmaceuticals in environmental and wastewater samples: a review. Anal Bioanal Chem. 2006;386:941–952.
- Hilton MJ, Thomas KV. Determination of selected human pharmaceutical compounds in effluent and surface water samples by high-performance liquid chromatography–electrospray tandem mass spectrometry. J Chromatogr A. 2003;1015:129–141.
- Kaluzna-Czaplinska J. Current Applications of Gas Chromatography/Mass Spectrometry in the Study of Organic Acids in Urine. Crit Rev Anal Chem. 2011;41:114–123.
- Chandra P, Rathore AS, Lohidasan S, et al. Application of HPLC for the Simultaneous Determination of Aceclofenac, Paracetamol and Tramadol Hydrochloride in Pharmaceutical Dosage Form. Sci Pharm. 2012;80:337–351.
- Sanghavi BJ, Srivastava AK. Simultaneous voltammetric determination of acetaminophen and tramadol using Dowex50wx2 and gold nanoparticles modified glassy carbon paste electrode. Anal Chim Acta. 2011;706:246–254.
- Siddiqui MR, AlOthman ZA, Rahman N. Analytical techniques in pharmaceutical analysis: A review. Arab J Chem. 2013; doi.org/10.1016/j.arabjc.2013.04.016.
- Kosjek T, Heath E, Krbavcic A. Determination of non-steroidal anti-inflammatory drug (NSAIDs) residues in water samples. Environ Int. 2005;31:679–685.
- Gouda AA, Kotb-El-Sayed MI, Amin AS, et al. Spectrophotometric and spectrofluorometric methods for the determination of non-steroidal anti-inflammatory drugs: a review. Arab J Chem. 2013;6:145–163.
- Behera S, Ghanty S, Ahmad F, et al. UV-Visible Spectrophotometric Method Development and Validation of Assay of Paracetamol Tablet Formulation. J Anal Bioanal Technigues. 2012;3:151–156.
- Alghamdi AF. Voltammetric Analysis of Montelukast Sodium in Commercial Tablet and Biological Samples Using the Hanging Mercury Drop Electrode. Port Electrochim Acta. 2014;32:51-64.
- Tyszczuk-Rotko K, Beczkowska I, Wójciak-Kosior M, et al. Simultaneous voltammetric determination of paracetamol and ascorbic acid using a boron-doped diamond electrode modified with Nafion and lead films. Talanta. 2014;129:384–391.
- Ziyatdinova G, Budnikov H. Electroanalysis of antioxidants in pharmaceutical dosage forms: state-of-the-art and perspectives. Monatsh Chem. 2015;146:741–753.
- Mehta JP, Pandya KI, Godhani DR. Electrochemical Investigation of Divalent State of Praseodymium and Samarium in Non-aqueous Medium. Port Electrochim Acta. 2015;33:183–194.
- Silvia MLS, Garcia MBQ, Lima JLFC, et al. Flow system with electrochemical detection for determination of paracetamol in pharmaceutical preparations. Port Electrochim Acta. 2006;24:261–271.
- Habibi B, Jahanbakhshi M, Pournaghi-Azar MH. Differential pulse voltammetric simultaneous determination of acetaminophen and ascorbic acid using single-walled carbon nanotube-modified carbon-ceramic electrode. Anal Biochem. 2011;411:167–175.
- Sanghavi BJ, Srivastava AK. Simultaneous voltammetric determination of acetaminophen, aspirin and caffeine using an in situ surfactant-modified multiwalled carbon nanotube paste electrode. Electrochim Acta. 2010;55:8638–8648.
- Chrzescijanska E, Wudarska E, Kusmierek E, et al. Study of acetylsalicylic acid electroreduction behavior at platinum electrode. J Electroanal Chem. 2014;713:17–21.
- Masek A, Chrzescijanska E, Zaborski M. Voltammetric and FTIR Spectroscopic Studies of the Oxidation of Retinyl Propionate at Pt Electrode in Non-Aqueous Media. Int J Electrochem Sci. 2014;9:6809–6820.
- Babaei A, Taheri AR, Afrasiabi M. A Multi-Walled Carbon Nanotube-Modified Glassy Carbon Electrode as a New Sensor for the Sensitive Simultaneous Determination of Paracetamol and Tramadol in Pharmaceutical Preparations and Biological Fluids. J Braz Chem Soc. 2011;22:1549–1558.
- Santos AL, Takeuchi RM, Stradiotto NR. Electrochemical, Spectrophotometric and Liquid-Chromatographic Approaches for Analysis of Tropical Disease Drugs. Curr Pharm Anal. 2009;5:69–88.
- Masek A, Chrzescijanska E, Zaborski M. Characteristics of curcumin using cyclic voltammetry, UV–vis, fluorescence and thermogravimetric analysis. Electrochim Acta. 2013;107:441–447.
- Radi AE, Khafagy A, El-shobaky A, et al. Anodic Voltammetric determination of gemifloxacin using screen-printed carbon electrode. J Pharm Anal. 2013;3:132–136.
- Masek A, Chrzescijanska E, Kosmalska A, et al. Antioxidant activity determination in Sencha and Gun Powder green tea extracts with the application of voltammetry and UV-VIS spectrophotometry. C R Chimie. 2012;15:424–427.
- Silva TA, Zanin H, Vicentini FC, et al. Electrochemical determination of rosuvastatin calcium in pharmaceutical and human body fluid samples using a composite of vertically aligned carbon nanotubes and graphene oxide as the electrode material. Sens Actuators B: Chem. 2015;218:51–59.
- Teófilo RF, Reis EL, Reis C, et al. Glyphosate Determination in Soil, Water and Vegetables Using DPV Optimized by Response Surface Methodology. Port Electrochim Acta. 2008;26:325–337.
- Wudarska E, Chrzescijanska E, Kusmierek E, et al. Voltammetric studies of acetylsalicylic acid electrooxidation at platinum electrode. Electrochim Acta. 2013;93:189–194.
- Hotta H, Nagano S, Ueda M, et al. Higher radical scavenging activities of polyphenolic antioxidants can be ascribed to chemical reactions following their oxidation. Biochim Biophys Acta. 2002;1572:123–132.
- Masek A, Chrzescijanska E, Zaborski M, et al. Characterisation of the antioxidant activity of riboflavin in an elastomeric composite. C R Chimie. 2012;15:524–526.
- Masek A, Chrzescijanska E, Zaborski M. Electrochemical Properties of Catechin in Non-Aqueous Media. Int J Electrochem Sci. 2015;10:2504–2514.
- Wudarska E, Chrzescijanska E, Kusmierek E, et al. Voltammetric study of the behaviour of N-acetyl-p-aminophenol in aqueous solutions at a platinum electrode. C R Chimie. 2015;18:993–1000.
- Wudarska E, Chrzescijanska E, Kusmierek E. Electroreduction of Salicylic Acid, Acetylsalicylic Acid and Pharmaceutical Products Containing these Compounds. Port Electrochim Acta. 2014;32:295–302.
- Feng X, Zhang Q, Cong P, et al. Determination of the paracetamol degradation process with online UV spectroscopic and multivariate curve resolution-alternating least squares methods: comparative validation by HPLC. Anal Methods. 2013;5:5286–5293.
- Aguilar CA, Montalvo C, Ceron JG, et al. Photocatalytic Degradation of Acetaminophen. Int J Environ Res. 2011;5:1071–1078.
- Delvadiya K, Kimbahune R, Kabra P, et al. Spectrophotometric simultaneous analysis of paracetamol, propyphenazone and caffeine in tablet dosage forms. Int J Pharm Sci. 2011;3:170–174.
- Engin C, Yilmaz S, G. Saglikoglu G, et al. Electroanalytical Investigation of Paracetamol on Glassy Carbon Electrode by Voltammetry. Int J Electrochem Sci. 2015;10:1916–1925.
- Braggio S, Barnaby RJ, Grossi P, et al. A strategy for validation of bioanalytical methods. J Pharm Biomed Anal. 1996;14:375–388.
- Gouda AA, Hassan WS. Spectrophotometric determination of etodolac in pure form and pharmaceutical formulations. Chem Centr J. 2008;2:7–14.
- ICH Harmonized Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2(R1) Geneva 2005.
- Kalambate PK, Biradar MR, Karna SP, et al. Adsorptive stripping differential pulse voltammetry determination of rivastigmine at graphene nanosheet-gold nanoparticle/carbon paste electrode. J Electroanal Chem. 2015;757:150–158.
- Jainn R, Yadav RK, Rather JA. Voltammetric quantitation of nitazoxanide by glassy carbon electrode. J Pharm Anal. 2013;3:452–455.
Received June 7, 2017; accepted 18 May, 2018
ENDEREÇO PARA CORRESPONDÊNCIA E-mail address: ewa.chrzescijanska@p.lodz.pl

{kind=link}
{kind=link}
{kind=link}