










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.36 no.5 Coimbra set. 2018
https://doi.org/10.4152/pea.201805325
A Kinetic Study of Oxalic Acid Electrochemical Oxidation on a Manganese Dioxide Rotating Cylinder Anode
Ali H. Abbara and Ammar S. Abbasb,*
a Chemical Engineering Department, University of Al-Qadisiyah, Iraq
b Chemical Engineering Department, University of Baghdad, Iraq
Abstract
This paper deals with the theory and kinetics of oxalic acid electrochemical oxidation, in an acidic solution containing sodium chloride, using a manganese dioxide rotating cylinder anode. Voltammetric and galvanostatic electrolysis techniques were used. The voltametric study shows a higher anodic wave corresponding to chlorine oxidation on the MnO2 electrode, prevailing oxalic acid indirect oxidation. Galvanostatic electrolysis studies confirmed that the rate constant is affected by chloride concentration, current density, agitation and temperature. Electrochemical oxidation rate was found to be a pseudo-first order kinetic process. A strongly linear relationship between the rate constant and chloride concentration was observed, while polynomial relations, with respect to current density and temperature, were found. The activation energy was found to be 14.541 kJ/mol, which suggests a diffusion control kinetic step in oxalic acid degradation. The findings of the present research validate that oxalic acid incineration can be successfully carried out on a MnO2 anode, in NaCl presence.
Keywords: electrochemical incineration; oxalic acid; manganese dioxide; sodium chloride; pseudo-kinetics and activation energy.
Introduction
Oxalic acid (OA) is one of the most toxic organic compounds. OA has several manufacturing and industrial uses, including: printing and dyeing fabrics; straw hats bleaching; paint removing; varnish; rust or ink stains; and wood cleaning. After these industrial processes, this compound is discharged in the water effluents. OA also is the main and final intermediate for the catalytic and electrochemical oxidation of several other organic compounds [1]. Therefore, OA incineration should be done before effluents are discharged into the environment.
Electrochemical technology has shown to be a promising method for the incineration of organic pollutants, such as OA, existing in wastewater [2]. The main reagent here are electrons, which are a clean reagent; consequently, there is no need for adding an extra reagent. Also, the electrochemical process high selectivity prevents the production of unwanted by-products. The reaction is usually carried out at room temperature and at a normal pressure [3].
Several anodic materials have been tested, but most of them presented substantial disadvantages, such as rapid loss of activity (graphite), high cost (Si-boron- doped diamond (BDD)), release of toxic ions (PbO2), limited service life (SnO2) and an incomplete oxidation (IrO2) [4, 5]. Therefore, a considerable effort is devoted to investigate cheaper anode materials with comparable performances, to be used in the treatment of wastewater containing organic compounds. Among these, manganese oxides (MnOx) materials represent an attractive family for electrodes manufacture, and they have been successfully used in wastewater treatments and other applications [6-9]. These oxides display excellent electrocatalytic properties, low cost, availability, environmental compatibility and chemical stability [6, 7]. Electrodes based on manganese oxide thin films may be obtained by different methods, such as sol-gel [10], physical vapor deposition [11], electrostatic spray deposition [12], electrochemical deposition [13], and chemical bath deposition [14, 15]. The low costs of equipment and the accurate control of the deposited thickness make the electrodeposition method very widely used in manganese dioxide synthesis [13].
In the electrochemical process, pollutants are destroyed by either a direct or indirect oxidation process. It has been generally observed that the direct oxidation of organic compounds at the electrode surface is very difficult, and that the rate of reaction is too slow [4, 16]. Bonfatti et al. [16] showed that OA electrochemical oxidation could be carried out with higher current efficiency at Pt, in the presence of suitable amounts of NaCl. A higher chemical oxygen demand (COD) removal rate was observed when NaCl was added at a concentration of 1 g/L in OA electrochemical incineration, using a rotating MnO2 anode [7]. Furthermore, pseudo-first-order reaction kinetics was reported for the degradation of various organic compounds, with chloride as a supporting electrolyte [17, 18].
Electrochemical oxidation mechanism
For both direct and indirect carboxylic acids electrochemical oxidation on metal oxide anodes, a generalized degradation mechanism has been proposed by Comminilles [19]. During electrolysis, water molecules dissociate to form an OH radical with the loss of an electron, and the radical is simultaneously adsorbed onto the metal oxide.
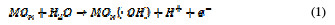
This is termed as physisorbed active oxygen. In the second step, the adsorbed hydroxide radicals react with the oxygen already present in the metal oxide anode, causing the transition of oxygen from the adsorbed hydroxyl radical to the lattice of the oxide anode, to form a higher oxide.
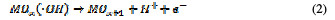
This is called as chemisorbed active oxygen, which reacts with carboxylic acids and converts them into CO2 and other intermediate products. The formed products undergo further oxidation in a similar way; thereby, complete oxidation takes place. Comminilles [19] also reported that chemisorbed active oxygen (MOx+1) is involved in the formation of particular oxidation products. If there is no oxidizable organic compound present in the medium, active oxygen on the oxide anode of both physisorbed and chemisorbed will liberate dioxygen, according to equations (3) and (4).
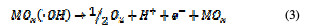
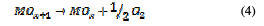
In the same way, an indirect oxidation mechanism has been proposed for the oxide anode, with chloride as a supporting electrolyte for the removal of organic pollutants from waste water [20]:
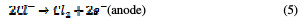
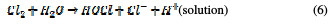
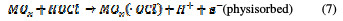
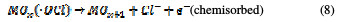
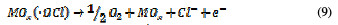

Physisorbed •OCl radicals and chemisorbed active oxygen participate in the organic compound degradation.
According to equations (9) and (10), in the absence of any oxidizable organics, the active oxidative species produce dioxygen and chloride ions. In acidic conditions, chlorine is the main oxidative agent for organic matter oxidation. In alkaline solutions, OCl-, ClO3- and free hydroxyl radicals are efficient oxidative agents [21]. The direct oxidation of organic compounds and its attack by the hydroxyl radical are negligible, in comparison with the attack by chlorine radicals.
Although some works are available for OA electrochemical oxidation using different anodic materials [7, 22], research on OA kinetic incineration using a MnO2 anode is not available. In the present work, a study of OA incineration kinetics by indirect electrochemical oxidation with a MnO2 electrode is reported.
Material and methods
All chemicals used in the experiments were of reagent grade, and there was no need for further purification. MnSO4.H2O (Thomas Baker), H2SO4 (Riedel- deHaën), OA (LOBA Chemie), NaCl (Thomas Baker) and distilled water were used in the preparation of all aqueous solutions.
Manganese dioxide was electroplated on a graphite rod of 1.98 cm diameter and a 32.3 cm2 active surface area, by anodic deposition on an acidified sulfate bath (100 g/L MnSO4- 90 g/L H2SO4). Details of experimental and used apparatus were described in a previous work [7].
Cyclic voltammetries were performed in a conventional three-electrode cell (Pyrex® glass) at room temperature. A manganese dioxide rotating cylinder electrode, with a submerged area of 12.3 cm2 rotated at 200 rpm, was used as working electrode. A platinum plate (0.35 cm2) and a saturated calomel electrode were used as counter and reference electrode, respectively. Electrochemical experiments were controlled with a potentiostat PGSTAT30 (Autolab) at a scan rate of 100 mV/s. Potential values referred to a saturated calomel electrode (SCE).
OA bulk oxidation was performed in a single-body, thermostated Pyrex glass cell of 0.75 liter; the anode was a MnO2 rotating cylinder electrode, with a submerged area of 32.3 cm2, and the supporting electrolyte was 0.5 M Na2SO4 containing 0.1 M OA. The final pH value was 2, adjusted by adding a 1 M NaOH or H2SO4 solution. The pH was kept constant at this value during electrolysis. The total volume of the electrolyte was 0.6 liter at each run. A hollow nickel cylinder with an inside diameter of 8 cm and length of 12 cm was used as counter electrode. The galvanostatic experiments of OA electro-oxidation were performed using a DC power supply (UNI-T: UTP3315TF-L). The effect of NaCl concentration in the range of 1 to 5g/L was investigated at a constant current density of 30 mA/cm2, while current densities in the range of 10 to 60 mA/cm2 were applied for studying the effect of current density (I) on the reaction rate constant. The effect of electrode rotation speed (Ï) was investigated in the range of 0 to 400 rpm using an electrical gearbox stirrer (Heidolph) with an electronic tachometer. Furthermore, temperature effect (T) was investigated in the range from 25 to 60 °C. During the electrolysis, OA residual content was determined using a conventional titration method with KMnO4 [22].
Results and discussion
Anode cyclic voltammograms performance
In order to have some preliminary information on the reactivity of active chlorine with oxalic acid, some cyclic voltammograms were recorded with and without NaCl and OA addition at the MnO2 anode in acidic conditions. As shown in Fig. 1, the addition of NaCl to a water solution produces a higher anodic wave.
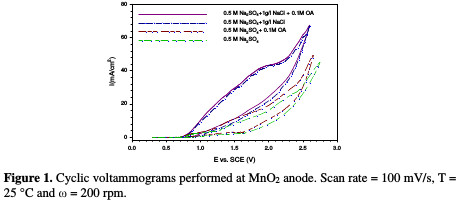
According to literature, such behavior can be attributed to the oxidation of Cl ions to Cl2 [23]. The same cyclic voltammetry behavior was obtained by Scialdone et al. [24], when OA, at a concentration of 5 mM, was incinerated on an IrO2-Ta2O5 anode; however, the anodic wave in the present work was higher, due to the used OA higher concentration.
Results of the previous work [7] showed that NaCl addition led to an increase in COD removal percentage from 26.40 %, in the case of free NaCl, to 79.50 %, with the addition of 1 g/L of NaCl. This increase in COD removal is an indication of the effective role of NaCl during OA indirect oxidation, when the MnO2 anode was used, as a result of chlorine overpotential on this electrode, which enables more chlorine molecules to attack OA.
Indirect electrochemical oxidation kinetics
In indirect electrochemical oxidation, OA removal rate is proportional to OA concentration, and also to chlorine/hypochlorite concentration, since the latter mediated indirect oxidation. OA removal rate can be written as:

Electrochemical treatment involves the application of an electric current to the effluent, to convert chloride to chlorine and hypochlorite. Chlorine and hypochlorite will oxidize OA, and be reduced to a chloride ion. The process is then repeated in a cyclic way. Chlorine/hypochlorite concentration during electrolysis is assumed to be constant, and so, equation (11) can be written as a pseudo-first order kinetics equation.

The integration of equation (12) leads to a natural logarithmic relation between OA concentration and time, as follows,
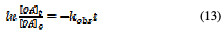
which could be written as an exponential decay function:

By using non-linear least squared fittings (exponential decay) of the untransformed original equation (Eq. (14)), the observed rate constant kobs (min-1) value can be estimated [25].
Effect of NaCl concentration on OA removal
Fig. 2 illustrates the decrease in normalized OA concentration, [OA]t/[OA]o, plotted against electrolysis time, with their exponential decay fitting, using different NaCl concentrations.
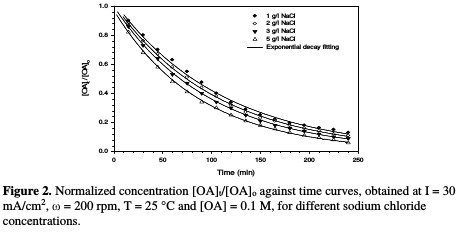
Table 1 and Fig. 3 show the rate constant against chloride concentration.
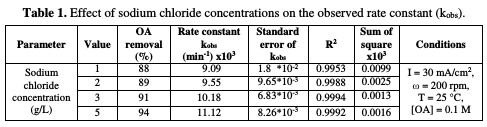
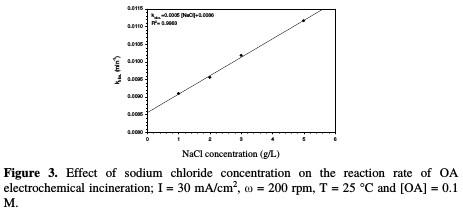
The increase in chloride concentration in the electrolytic solution leads to a higher reaction rate. A previous study [26] showed that an increase in chloride concentration reduced the overpotential required for chloride anodic oxidation, resulting in a higher production of hypochlorite. It is clear that there is a strong linearity between the rate constant and chloride concentration. The rate constant reported in Table 1 increased from 0.0091 to 0.011 min-1; when NaCl concentration rose from 1 to 5 g/L, the values of R2 varied between 0.9953 and 0.9994 for different chloride concentrations. Huang et al. [27] found the same linear relationship between the rate constant and chloride concentration when OA degraded, using a Ti-DAS net anode. OA removal efficiency increased from 88 to 94 %, as NaCl concentration increased from 1 to 5 g/L (Table 1). However, using a NaCl concentration higher than 3 g/L is not recommended, because the halogenated organic compound can generate more toxic materials than those of OA [24].
Effect of current density on OA removal
Fig. 4 shows the decrease in normalized OA concentration, [OA]t/[OA]o, plotted against electrolysis time, with their exponential decay fitting at different current densities.
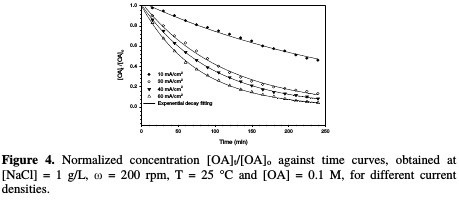
The rate constant against the current density is shown in Fig. 5 and Table 2.
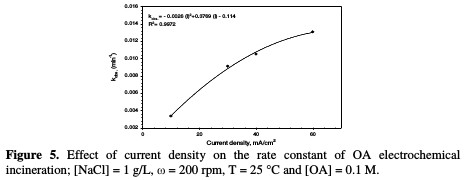
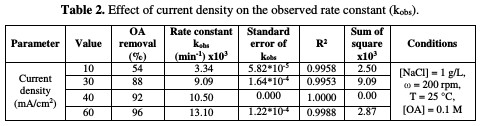
In this study, when the current density increases from 10 to 60 mA/cm2, the rate constant increases from 0.0033 to 0.0131 min-1. The relation between current density and the rate constant was fitted to be a polynomial. The enhancing effect of current density is attributed to the improvement of chlorine/hypochlorite production rate that enhances the indirect oxidation effect during electrolysis.
Huang et al. [26] found a linear relationship between the rate constant and the current density when OA degraded, using a Ti-DAS net anode in NaCl presence. According to data in Table 2, OA removal percentage increased from 54 to 96 %, as the current density increased from 10 to 60 mA/cm2. This result is an indication that the current density is the most important factor in OA incineration in the presence of chloride ions.
Effect of rotation speed on OA removal
The decrease in normalized OA concentration, [OA]t/[OA]o, plotted against electrolysis time, with their exponential decay fitting at different rotation speed, is presented in Fig. 6.
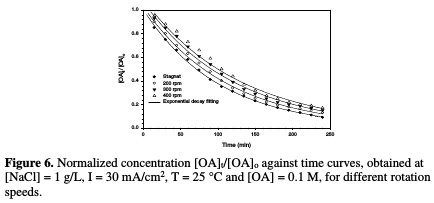
The relation between the rate constant and agitation is depicted in Table 3 and Fig. 7.
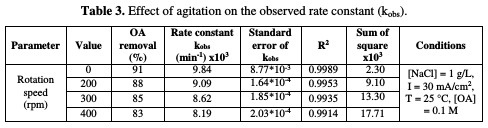
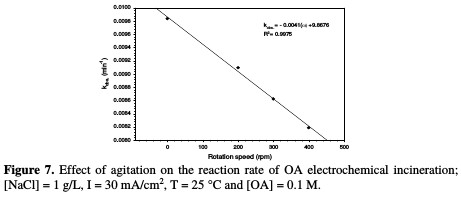
It is interesting to note that the absence of agitation gives higher rate constants, while increasing rotation speed more than 200 rpm leads to a drastic drop in the reaction rate. These behaviors may be interrupted, as higher electrode rotation velocities would not enable the reaction of chlorine with OA, and a part of the formed chlorine would be liberated to escape outside the cell; hence, a lower reaction rate takes place. It is observed that there is a substantial inverse linearity between the rate constant and agitation.
Effect of temperature on OA removal and reaction kinetics The decrease in normalized OA concentration, [OA]t/[OA]o, plotted against electrolysis time, with their exponential decay fitting at different temperatures, is shown in Fig. 8.
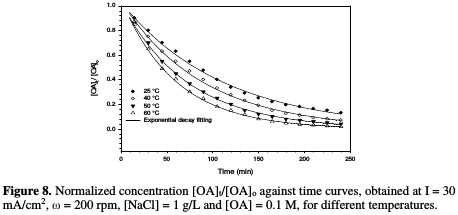
The relation between the rate constant and temperature is illustrated in Fig. 9 and Table 4.
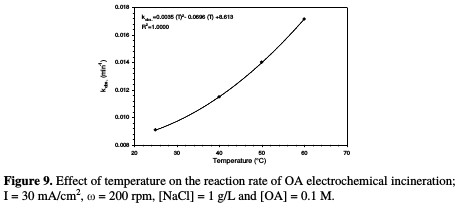
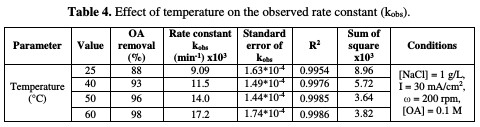
A polynomial relationship between the rate constant and temperature was observed. The effect of temperature on OA anodic incineration was widely investigated by the group of De Battisti [22], in the presence of several anodes, Scialdone et al. [28], for the case of DSA-O2 anodes, and Canizares et al. [29], for BDD. According to literature data, the temperature effect on OA degradation strongly depends on the anodic material nature. With BDD, a temperature increase gave rise to a slight change in OA abatement [22, 29], while a drastic increase at IrO2 based anodes was observed [22, 28]. In the present system, a drastic increase in OA abatement was observed in the presence of NaCl. Therefore, MnO2 has a catalytic activity towards OA, which increased with higher temperatures.
The activation energy was calculated from the study of the temperature effect on the rate constant (Arrhenius' law), considering that the activation energy range of a typical diffusion-controlled process is less than 20 kJ/mol [30]. Fig. 10 shows the relation between ln kabs and the reciprocal of temperature (Arrhenius' plot).
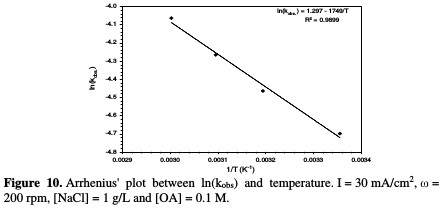
The data allowed estimating an activation energy of 14.541 kJ/mol, which confirms that the present system is limited by mass transfer [31].
Conclusion
The present research confirmed that the rate constant of OA degradation reaction is directly proportional to the increase in NaCl concentration, current density, and temperature, while it is inversely proportional to the increase in the anode rotation speed. Two important points are worthy of attention: firstly, current density has a significant role on OA incineration in the present research, where a higher reaction rate can be achieved; secondly, temperatureâs influence shows a positive behavior, according to Arrheniusâ law, in the current work, while an adverse effect was observed with other organic compounds, such as glucose [32], or no significant effect, in the case of anodes such as BDD [29]. This discrepancy between the present work and previous works may be attributed to the role of active chlorine in the electrochemical incineration, and to the catalytic activity of the MnO2 anode. Results of activation energy suggested that the reaction is controlled by the species diffusion towards the anode surface, and by their reaction with the active chlorine, while direct OA oxidation at the electrode surface is insignificant.
References
1. Chollier MJ, Epron F, Lamy-Pitara E, et al. Catalysis Today. 1999;18:291. [ Links ]
2. Xiong Y, Karlsson H. Adv Environ Res. 2002;7:139. [ Links ]
3. Raijeshwar K, Ibanez JG, Swain GM. J Appl Electrochem. 1994;24:1077. [ Links ]
4. Martinez-Huitle CA, Ferro S. Chem Soc Rev. 2006;35:1324. [ Links ]
5. Panizza M, Cerisola G. Electrochim Acta. 2005;51:191. [ Links ]
6. Sotgiua G, Foderà b M, Marrab F, et al. Chem Eng Trans. 2014;41:115.
7. Abbar AH, Salman RH, Abbas AS. J Electrochem Soc. 2016;163:E333. [ Links ]
8. Fergus JW. J Power Sources. 2010;195:939. [ Links ]
9. Wei W, Cui X, Chen W, et al. Chem Soc Rev. 2011;40:1697. [ Links ]
10. Chen CY, Wang SC, Tien YH, et al. Thin Solid Films. 2009;518:1557. [ Links ]
11. Djurfors B, Broughton JN, Brett MJ, et al. Acta Mater. 2005;53:957. [ Links ]
12. Nam KW, Kim KB. J Electrochem Soc. 2006;153:A81. [ Links ]
13. Chou SL, Cheng FY, Chen J. J Power Sources. 2006;162:727. [ Links ]
14. Xia H, Xiao W, Lai MO, et al. Nanoscale Res Lett. 2009;4:1035. [ Links ]
15. Xia H, Lai MO, Lu L. JOM. 2011;63:49. [ Links ]
16. Bonfatti F, Battisti A, Ferro S, et al. Electrochem Acta. 2000;46:305. [ Links ]
17. Rajkurnae D, Palanivelu K, Mohan N. Ind J Chem Technol. 2003;10:396. [ Links ]
18. Szpyrkowiez L, Juzzolino C, Kaul SN, et al. Ind Eng Chem Res. 2000;39:3241. [ Links ]
19. Cominiellis C. Electrochem Acta. 1994;39:1857. [ Links ]
20. Buso A, Balbo L, Giomo M, et al. Ind Eng Chem Res. 2000;39:494. [ Links ]
21. Deborde M, Von Gunten U. Water Res. 2008;42:13. [ Links ]
22. Martinez-Huitle CA, Ferro S, De Battisti A. Eletrochim Acta. 2004;49:4027. [ Links ]
23. Szpyrkowicz L, Radaelli M, Daniele S. Catalysis Today. 2005;100:425. [ Links ]
24. Scialdone O, Randazzo S, Galia A, Silvestri G. Water Res. 2009;43:2260. [ Links ]
25. Lente G. Deterministic Kinetics in Chemistry and Systems Biology. London: Springer; 2015. [ Links ]
26. Mohan N. Studies on electrochemical oxidation of acid dye effluent (PhD thesis). Chennai: Anna University; 2000. [ Links ]
27. Huang Y-H, Shih Y-J, Liu C-H. J Hazard Mater. 2011;188:188. [ Links ]
28. Scialdone O, Randazzo S, Galia A, et al. Electrochim Acta. 2009;54:1210. [ Links ]
29. Canizares P, Garcia-Gomez J, Lobato J, et al. Ind Eng Chem Res. 2003;42:956. [ Links ]
30. Ferreira M, Pinto MF, Soares OSGP, et al. Electrochim Acta. 2012;60:278. [ Links ]
31. Benson SW. The Foundations of Chemical Kinetics. New York: McGraw-Hill; 1960. [ Links ]
32. Bonfatti F, Ferro S, Lavezzo F, et al. J Electrochem Soc. 2000;147:592. [ Links ]
Acknowledgements
The authors wish to acknowledge the helpful suggestions and gracious technical assistance from the staff of the Chemical Engineering Department, College of Engineering, University of Baghdad, Iraq.
*Corresponding author. E-mail address: ali.abbar@qu.edu.iq
Received February 17, 2017; accepted November 10, 2017

