










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.35 no.3 Coimbra maio 2017
https://doi.org/10.4152/pea.201703179
Corrosion of a Ni-Al Composite Coating in 2 M NaCl Solution
Ikenna B. Onyeachua , Demian I. Njokub , Emeka E. Oguzieb,* and X. Pengc
a Department of Chemistry, Edo University Iyamho, Edo State, Nigeria
b Electrochemistry and Material Science Research Unit (EMRU), Department of Chemistry, Federal University of Technology Owerri, Imo State, Nigeria
c State Key Laboratory for Corrosion and Protection, Institute of Metal Research, Chinese Academy of Science, 62 Wencui Road, Shenyang 110016, China
Abstract
A Ni-Al composite was electrodeposited on a Ni substrate, and its corrosion behavior was observed in 2 M NaCl solution, compared with a pure Ni coating. The Al particles increased the porosity of the composite and encouraged charge percolation, both at the corrosion product layer-solution interface and at the substrate-solution interface, based on EIS characterization. This phenomenon greatly decreased the corrosion potential, and increased both cathodic and anodic current densities in the active region, as well as the passive current density in the passive potential range, during polarization of the composite. Although a continuous Al3+ ions supply to the passivation front was suspected, based on the longer passivation potential of the Ni-Al composite, the simultaneous consumption of the Al products by the chloride ions is the reason for serious cracking and localized collapse of the composite corrosion layer, as confirmed by SEM. This conferred lower corrosion resistance on the Ni-Al composite, compared to the pure Ni coating, in the 2 M NaCl solution.
Keywords: Ni-Al composite, passive layer, EIS, SEM, corrosion resistance.
Introduction
The reinforcement of the pure nickel (Ni) coating with micrometer or nanometer-size second phase particles, to form a Ni matrix composite, is a veritable approach to improve the material and corrosion characteristics of the Ni coating [1-7]. A Ni-Al composite consists of a Ni coating reinforced with an aluminum (Al) particle. The electrodeposition of a Ni-Al composite commonly involves the deliberate loading of a Ni deposition bath with a given amount of Al particles, so that, assisted by a means of agitation, the Ni2+ ion adsorbs on the particle and transports it to the substrate, where it becomes entrapped during the Ni2+ reduction [8, 9]. Upon annealing treatment of the electrodeposited Ni-Al composite, the Al particle usually reacts with the Ni matrix, and the process is an important step towards the fabrication of an γ-Ni3Al intermetallic ultrafine-grain, a potential structural material well known for its low density, high strength at elevated temperature and higher resistance to high temperature oxidation [10-12] and electrochemical corrosion [13], compared with the polycrystalline γ-Ni3Al counterpart.
Despite refining the Ni grain size and morphology, which is important for increased corrosion resistance, it is expected that the Al particle in a Ni-Al composite could also augment the Ni corrosion resistance by enriching the Ni corrosion product layer with Al2O3. The formation of Al2O3 is fundamental for the high corrosion resistance of many materials containing Al. Nevertheless, in aqueous solutions containing chloride ions, Al2O3 itself can be stealthily destroyed through its transformation into soluble chloride complexes [14-16]. In a previous work [17], we complemented electrochemical techniques with surface probe and depth profiling techniques, and reported that, due to the ability of the Al2O3 to acquire surface hydration and become gradually transformed into the less protective Al(OH)3, an incubation time was required for the formation of a well-developed Al2O3 layer, which increased the corrosion resistance of a Ni-Al composite coating, compared to a pure Ni coating, in a neutral 3.5% NaCl solution. Conversely, the Al2O3 was greatly unstable and consumed in a more aggressive 3.5% NaCl solution acidified with 0.05 M H2SO4 solution, thus, decreasing the Ni-Al corrosion resistance [18]. In order to fully appraise the role of the Al particle during electrochemical corrosion of a Ni-Al composite in chloride-containing solutions, an important approach would also be to investigate this phenomenon in a strongly saline solution with higher chloride ion concentration.
In the present work, we characterize the corrosion behavior of an electrodeposited Ni-Al composite, compared with a pure Ni coating, in 2 M NaCl solution. We focus on the electrochemical characteristics and the surface probe of the corrosion product layers formed by the coatings.
Experimental
All reagents used in this work were analytical (BDH) grade chemicals used as source without further purification. All solutions were prepared with deionized water. The Ni coating and Ni-Al composite were fabricated on 99.9% Ni substrates with dimensions (12×10×12) mm3, using direct-current electrodeposition. The preparation of the Ni substrates involved mechanical abrading to final 800 grit size, using SiC paper, washing with distilled water, ultrasonic cleaning in acetone and, thereafter, drying with a mechanical dryer. The procedure for the Ni and Ni-Al electrodeposition has been detailed in our earlier reports [17, 18]. The average content of the co-deposited Al particle was found to be 28wt%. The as-deposited coatings were dried in an oven at 105 oC for 2 h, and kept in an oxygen-free desiccator. The electrochemical corrosion behavior of the Ni coating and Ni-Al composite was observed in 2 M NaCl solution at room temperature, using electrochemical impedance spectroscopy (EIS) and potentiodynamic polarization techniques, with the aid of a PARSTAT 273A Potentiostat/Galvanostat (Princeton Applied Research) connected to a signal recovery model 5210 lock-in amplifier for EIS acquisition. The coatings were embedded in a mixture of paraffin and rosin, so that an area of 100 mm2 was exposed for the corrosion characterization. The prepared coatings were deployed as working electrodes in a three-electrode system, wherein a platinum sheet functioned as counter electrode, and a saturated calomel electrode (SCE) was the reference electrode. The working electrodes were first allowed to attain open circuit potential (OCP) before the electrochemical characterization. For the EIS measurement, the coatings were subjected to a signal amplitude perturbation of 10 mV through a frequency range of 100 kHz to 0.1 Hz. Potentiodynamic polarization measurement was performed by applying 0.166 mV/s potential scan between -0.25 V/OCP and +0.40 V/SCE. The surface morphology of the Ni coating and Ni-Al composite was visualized, before and after polarization, using the SEM (FEI Inspect/OXFORDINSTRUMENTS-X-Max) hyphenated with an energy dispersive X-ray (EDAX) spectrophotometer for the acquisition of the elemental composition.
Results and discussion
EIS characterization
The EIS results obtained for the pure Ni coating and the Ni-Al composite, at OCP in the 2 M NaCl solution, are expressed in Nyquist, Bode phase angle and Bode absolute impedance formats, and shown in Fig. 1.
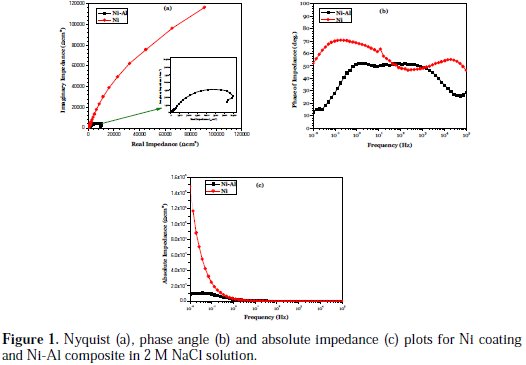
In Fig. 1(a), the Nyquist plots showed curves of incomplete semi-circles: a slight semi-circle at high frequency and a larger one at low frequency. The size of the Nyquist plot usually correlates with the corrosion resistance of the material in a corrosion solution [19-22]. The clearly larger size of the Nyquist plot exhibited by the pure Ni coating implies higher corrosion resistance compared with the Ni-Al composite. In the Bode phase angle plots, Fig. 1(b), both coatings displayed two impedance maxima, Fig. 1(c), which imply that two time constants could be employed to explain the electrochemical processes occurring at the surfaces of the Ni and Ni- Al coatings in 2 M NaCl solution. Such two time constants usually represent impedance processes like charge transfer, mass transfer and film effects occurring at a corrosion product layer-solution interface and a substrate-solution interface beneath the corrosion product layer [23]. The high frequency phase angle maximum in EIS characterization is commonly associated with resistance to phenomena, such as uptake of water and intrusion of electrolyte salt, whereas the low frequency maximum concerns resistance to substrate-controlled processes including localized defects [23]. The pure Ni coating displayed higher phase angle maxima throughout the frequency scan compared with the Ni-Al composite, an attestation to the higher corrosion resistance of the former in the 2 M NaCl solution. By fitting the impedance data with the ZSimpWin software, Fig. 2 shows the most suitable equivalence circuit to model the electrochemical corrosion mechanism of the Ni coating and Ni-Al composite in 2 M NaCl solution.
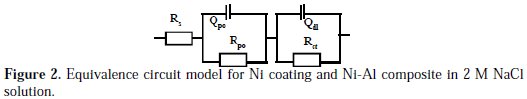
The values of the electrical elements in the equivalence circuit are detailed in Table 1.
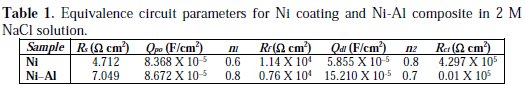
Rs is the solution resistance between the reference and working electrodes; Qpo and Rpo, respectively, stand for the capacitance of a corrosion product layer and its resistance to transfer of ions; and Qdl and Rct, respectively, represent the capacitance of the double layer and resistance to charge transfer in the regions beneath the corrosion product layer. The Qdl for the Ni-Al composite was approximately three times as high as that of the Ni coating, which implies greater solution percolation within the composite.
More so, the Rct, which is a measure of the corrosion resistance of the coatings, was far lower for the composite than for the Ni coating. Thus, the Al particle increased the porosity of the Ni coating, which promoted the 2 M NaCl solution penetration through the numerous micro pores on the composite surface. Based on the surface hydration characteristic well known for the Al2O3, as reported elsewhere [21], the solution accumulation in the composite interior should, therefore, favor its transformation into Al(OH)3 which, in turn, is rapidly consumed in the presence of high chloride concentration in the 2 M NaCl solution. The Al2O3 to Al(OH)3 transformation, in the presence of water and subsequent dissolution by chloride ions, was also reported [24, 25]. This explains the lower corrosion resistance of the Ni-Al composite at open circuit potential, compared with the pure Ni coating, in 2 M NaCl solution.
Potentiodynamic polarization characterization
Potentiodynamic polarization curves derived for the Ni coating and Ni-Al composite in 2 M NaCl solution are presented in Fig. 3.
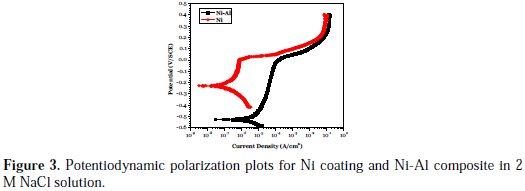
The values of the polarization parameters, such as corrosion potential (Ecorr) and corrosion current density (icorr), which were derived from the intersection of the cathodic and anodic Tafel slopes drawn ±5 mV around the transition potential, are given in Table 2.
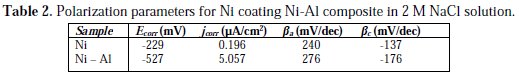
The polarization result shows that the Ni-Al composite displayed a strongly more negative Ecorr (-527 mV), compared with the pure Ni coating (-229 mV). This reveals greater propensity for the Ni-Al composite to become dissoluble in the 2 M NaCl solution. Furthermore, the icorr value of 5.057 μA/cm2 for the composite was far greater than the icorr value of 0.1958 μA/cm2 for the pure Ni coating.
The icorr value is a measure of the corrosion rate of the coatings. Thus, there is a higher rate of cathodic and anodic half-reaction occurring on the surface of the Ni-Al composite, compared with the Ni coating in 2 M NaCl solution. This strongly correlates with the mechanism of solution accumulation in the composite pores at open circuit (equilibrium) potential, as reported during EIS measurement. Away from the equilibrium potential during anodic polarization, both Ni coating and Ni-Al composite exhibited similar phenomena, namely: (i) an active region during early scan; (ii) a region where the applied potential caused only minimal increase in current density, and could be regarded as a passive region caused by a corrosion product layer formation; (iii) a region where the current density sharply increases with the applied potential, attributed to the initiation of serious localized phenomena, such as pitting corrosion; and (iv) a region whereby the coatings attempt a gradual transition to trans-passivation. Although the Ni-Al composite displayed higher current density than the Ni coating, in the passive region (which implies greater transport of charges through the Ni-Al corrosion product layer), surprisingly, the composite sustained its passive nature over a longer potential range. This strongly indicates that, during the polarization, an increased outward migration of Al3+ ions occurs from the composite interior through the corrosion product layer grain's boundaries to the reaction front for the sustenance of passivation (which prolongs the passive potential). However, the Al corrosion products are simultaneously consumed as they form (which results in higher current than that of the Ni coating in that region). This phenomenon provides more support for the strong surface hydration of the formed Al2O3 and its transformation into Al(OH)3, which is readily consumed by chloride ions in the 2 M NaCl solution.
SEM characterization
After polarization in 2 M NaCl solution, the electrodeposited Ni coating, Fig. 4(a), transformed such that the Ni corrosion products scattered on the surfaces of the Ni grains; however, with no clear linkage into a continuous layer.

At some points, obvious consumption of the Ni grains could be noticed. The surface of the as-deposited Ni-Al composite (where the Al particle preferentially incorporated within the Ni grain boundaries), Fig. 4(c), was transformed into a corrosion product layer, somewhat, smoother when compared with the Ni coating, Fig. 4(d). The layer was, nevertheless, characterized by serious cracks, which led to obvious collapse of some portions. It is important to notice that, apart from the regions where the layer collapsed, numerous holes could be seen at the triple junction of the cracks along the Ni-Al corrosion product layer, which is strongly attributed to the points of serious consumption by chloride ions in the 2 M NaCl solution. Since Al2O3 formation should predominantly occur at the Ni grain boundaries and assist in the linkage of the Ni corrosion product layer, given that the Al particle is mainly incorporated therein, it must be the Al products consumption by the chloride ion that vigorously destroys the grain boundary adhesion and locally causes the collapse of the layer portions. This Al product consumption by chloride ions can be verified from the EDAX acquisition of Cl and Na with high composition of Al and O (than Ni), at a crack along the corrosion product layer, Fig. 4(e). This is, therefore, the reason for the lower corrosion resistance of the Ni-Al composite, than that of the pure Ni coating, in 2 M NaCl solution.
Conclusion
The electrodeposited Ni-Al composite exhibited lower corrosion resistance in 2 M NaCl solution, compared with the pure Ni coating, because the Al particle increases the porosity of the Ni coating and encourages the solution penetration during corrosion. This leads to an increased rate of dissolution of the composite surface. The Al corrosion products are readily consumed in the highly concentrated chloride solution, leading to serious cracking and localized collapse of the composite corrosion product layer.
References
1. Shi L, Chufeng S, Gao P, et al. Appl Surf Sci. 2006;252:3591. [ Links ]
2. Li J, Sun Y, Sun X, et al. Surf Coat Technol. 2005;192:331. [ Links ]
3. Zimmerman AF, Palumbo G, Aust KT, et al. Mater Sci Eng A. 2002;328:137. [ Links ]
4. Hasannejad H, Shahrabi T, Jafarian M. Mater Corros. 2012;1. [ Links ]
5. Szczygiel B, Kolodziej M. Electrochim Acta. 2005;50:4188. [ Links ]
6. Feng Q, Li T, Teng H, et al. Surf Coat Technol. 2008;4137. [ Links ]
7. Peng X, Zhang Y, Zhao J, et al. Electrochim Acta. 2006;51:4922. [ Links ]
8. Susan DF, Barmak K, Marder AR. Thin Solid Films. 1997;307:133. [ Links ]
9. Guglielmi N. Electrochem Soc. 1972;119:1009. [ Links ]
10. Peng X, Li M, Wang F. Corros Sci. 2011;53:1616. [ Links ]
11. Zhou YB, Zhang HJ. Trans Nonferrous Metal Soc China. 2011;21:322. [ Links ]
12. Yang X, Peng X, Wang F. Corros Sci. 2008;50:3227. [ Links ]
13. Onyeachu BI, Oguzie EE, Njoku DI, et al. Int J Sci Technol. 2014;3:728. [ Links ]
14. McCafferty E. Corros Sci. 2003;45:1421. [ Links ]
15. Ensinger W, Lensch O, Matsutani T. et al. Surf Coat Technol. 2005;196:231. [ Links ]
16. Jafarzadeh K, Shahrabi T, Hosseini MG. Corros Sci Eng Technol. 2009;44:144. [ Links ]
17. Onyeachu BI, Peng X, Oguzie EE, et al. Port Electrochim Acta. 2015;33:69. [ Links ]
18. Onyeachu BI, Peng X, Oguzie EE, et al. Int Sci Technol. 2014;3:711. [ Links ]
19. Oguzie EE, Adindu CB, Enenebeaku CK, et al. Phys Chem C. 2012;116:13603. [ Links ]
20. Njoku DI, Ukaga I, Onyeachu BI, et al. Mol Liq. 2016;219:417. [ Links ]
21. Khaled KF. Corros Sci. 2010;52:2905. [ Links ]
22. M.A. Chidiebere, C.E. Ogukwe, and K.L. Oguzie et al., Ind. Eng. Chem. Res. 2012;15:668.
23. Y. Zhao, G. Wu, and H. Pan et al., Mater. Chem. Phy. 2012;132:187.
24. Chen C, Splinter SJ, Do T, et al. Surf Sci. 1997;382:L652. [ Links ]
25. Alwitt RS. In: Oxides and oxide films. Diggle JW, Vijh AK (eds.). New York: Marcel Dekker; 1976.
Acknowledgements
Onyeachu B. Ikenna is grateful to The World Academy of Sciences (TWAS) and the Chinese Academy of Sciences (CAS) for the award of a Postgraduate Fellowship.
*Corresponding author. E-mail address: emekaoguzie@gmail.com
Received August 20, 2016; accepted November 21, 2016

