










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.34 no.2 Coimbra mar. 2016
https://doi.org/10.4152/pea.201602085
Electrocatalytic oxidation of ethanol at silver chloride/ bromide modified carbon paste electrodes
Ghasem Karim-Nezhad* and Sara Pashazadeh
Department of Chemistry, Payame Noor University, PO BOX 19395-3697 Tehran, Iran
Abstract
Silver chloride modified carbon paste electrode was prepared as a new electrode and used to electrocatalytic oxidation of ethanol. For the first time, the catalytic oxidation of ethanol was demonstrated by cyclic voltammetry, chronoamperometry and amperometry methods at the surface of this modified carbon paste electrode. Compared to silver chloride modified carbon paste electrode, silver bromide modified carbon paste electrode and bare silver electrode catalysts, silver chloride modified carbon paste electrode exhibited markedly superior catalytic activity for the electrocatalytic oxidation of ethanol. It can be seen that the electrocatalytic efficiency of silver chloride modified carbon paste electrode is higher than the silver bromide modified carbon paste and bare silver electrodes. The catalytic oxidation peak current was linearly dependent on the ethanol concentration. The j0 for silver chloride modified carbon paste and silver bromide modified carbon paste electrodes are 11.2 and 5.4 folds respectively higher than that of the bare silver electrode. For silver chloride modified carbon paste electrode, the charge transfer coefficient (α), the number of electrons involved in the rate determining step (nα) and exchange current density (j0) were calculated as 0.46, 1 and 5.05×10-7 respectively. The modified electrode possesses high selectivity, good reproducibility and well stability.
Keywords: ethanol, modified carbon paste electrode, electrocatalytic oxidation, silver chloride, cyclic voltammetry.
Introduction
Low-carbon alcohols are currently viewed as one of the most promising renewable resources for replacing declining petrochemical reserves over the next decade [1]. Among the several small organic molecules, ethanol is one of the most promising fuels because of its low toxicity, abundant availability, low permeability (but not negligible) across proton exchange membrane [2] and higher energy density (1325.31 kJmol-1) than that of methanol (702.32 kJmol-1) [3]. Therefore, regarding the electro-oxidation of ethanol and the construction of a direct ethanol fuel cell (DEFC), intense research efforts have been seen in the past decades [4-6]. Recently the use of ethanol in electrocatalytic processes has received great attention in many research groups, justified by the development of more efficient and less polluting electrochemical energy conversion systems. Although metals such as Pt, Au, and Ag are very active in the anodic oxidation and cathodic reduction, they are too expensive for practical applications. The use of bare electrodes for electrochemical detection have a number of limitations, such as low sensitivity and reproducibility, slow electron transfer reaction, low stability over a wide range of solution composition and high overpotential at which the electron transfer process occurs. The chemical modifications of inert substrate electrodes with redox active thin films offer significant advantages in the design and development of electrochemical sensors. In operation, the redox active sites shuttle electrons between the analyte and the electrodes with significant reduction in activation overpotential [7].
Carbon paste electrode (CPE) is a special kind of heterogeneous carbon electrode consisting of mixture prepared from carbon powder (as graphite, glassy carbon and others carbonaceous materials) and a suitable water-immiscible or nonconducting binder [8]. The ease and speed of preparation and of obtaining a new reproducible surface, low residual current, porous surface, and low cost of carbon paste are some advantages of CPE over all other carbon electrodes. Therefore, CPE can provide a suitable electrode substrate for preparation of modified electrodes [9]. Modification of the paste matrix with various transition metal complexes [10-12] were reported in recent years. These electrodes have been widely used in electroanalysis due to their ability to catalyze the redox processes of some molecules of interest, since they facilitate the electron transfer [13]. A variety of mediators ranging from organic molecules to inorganic complexes such as prussian blue (PB) [14], polyoxometalates [15, 16] and ruthenium complexes [17] have been applied for the construction of CMCPE. Ag is one of important metals and can be used not only as modification metal but also as substrate metal in surface modification [18]. We previously proved that the presence of halides causes an increase activity in the electrocatalytic behavior of copper. The increased activity probably related to a more favorable adsorption of reactant or of intermediates leading to a higher surface concentration of electroactive molecules ready for being oxidized or it is due to the partial delocalization of the electronic density of reactant into the solid with possible consequent bond pre-dissociations which facilitates the oxidation or both [19, 20]. It seems that the presence of halides causes an increase activity in the electrocatalytic behavior of silver similar to copper.
The aim of this work is to develop a new modified carbon paste electrode using silver chloride and its application to electrocatalytic oxidation of ethanol in comparison to bare and silver bromide modified carbon paste electrodes.
Experimental
Reagents and solutions
Ethanol and other reagents were of analytical grade supplied by Merck (Darmstadt, Germany) and Sigma Aldrich and were used without further purification. Deionized water was used for the preparation of all solutions. All Electrochemical measurements were carried out in a conventional three-electrode cell powered by an electrochemical system comprising an AUTOLAB system with PGSTAT12 boards (ECO Chemie, Utrecht, and The Netherlands). The system was run on a PC using GPES 4.9 software. SC-MCP and SB-MCP electrode as working electrode (prepared as follows) were employed for the electrochemical studies. A platinum wire was employed as counter electrode and an Ag/AgCl electrode served as the reference electrode. All experiments were performed at room temperature of 25±2 °C.
Preparation of silver chloride/bromide modified carbon paste electrodes
The unmodified carbon paste electrode was prepared by mixing graphite powder with appropriate amount of mineral oil (paraffin) and thorough hand mixing in a mortar and pestle (70:30, w/w). Then a portion of the composite mixture was packed into the end of a polyethylene syringe (2.0 mm in diameter). Electrical contact was made by forcing a thin copper wire down into the syringe and into the back of the composite. The modified electrode was prepared by mixing unmodified composite with silver chloride (9.0%, w/w)/ silver bromide (11.0%, w/w). Finally, the modified composite was packed into the end of a polyethylene syringe. A fresh electrode surface was obtained by squeezing out a small portion of paste and polishing it with wet filter paper until a smooth surface was obtained.
Results and discussion
Electrochemical properties of the prepared SC-MCP electrode were investigated. For the activation of this electrode, the electrode was placed in 0.1 mol L-1 NaOH and the electrode potential was cycled between 170 and 800 mV (vs. Ag/AgCl) at a scan rate of 50 mVs-1 for 20 cycles in a cyclic voltammetry regime until a stable voltammogram was obtained (not shown). Results showed that with increase of the scan number, the currents for both anodic and cathodic peaks increase steadily for up to 20 runs. After 20 runs, the SC-MCP electrode shows reproducible cyclic voltamograms. A pair of redox peaks was observed which correspond to the couple Ag(II)/Ag(I) can be described by the reaction (1):
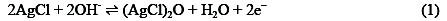
For SB-MCP electrode, similar voltammogram were obtained. The surface coverage can be evaluated from the equation Τ = Q/nFA, where Q is the charge obtained by integrating the anodic peak under the anodic wave of the cyclic voltammogram and other symbols have their usual meanings. Now by assuming the involvement of two electrons in the process, the calculated value of Î are 6.31×10-5 mol cm-2 for SC-MCP electrode and 3.26×10-7 mol cm-2 for SB-MCP electrode.
The voltammetric signals were affected by the composition of the paste. It was observed that the sensitivity of the sensor first rapidly increases with increasing the silver halide content in the paste up to about 9% SC-MCP and 11% SB-MCP, and then started to level off and even slightly decreases with the higher loadings (Fig.1).
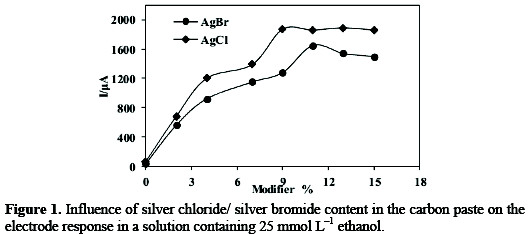
This is because the sites for adsorption increased with the increase of silver halide percentage in the modified electrode, while the excess of silver halide increase the resistance of the electrode. Higher concentrations (>9% SCMCP and 11% SB-MCP) showed a decrease in the peak current. This is presumably due to the reduction of conductive area at the electrode surface. Hence a silver halide (9% SC-MCP and 11% SB-MCP) modified carbon paste electrode was used throughout this work.
The cyclic voltammograms of modified carbon paste electrode (SC-MCP/ SBMCP) were recorded in different concentrations of NaOH solution containing 30 mmol L-1 ethanol (not shown). It is shown that, the high catalytic peak current is achieved above a NaOH concentration of 0.1 mol L-1. The results indicate that the OH- ion participates in the oxidation of ethanol and may also be adverse to the oxidation of ethanol for the competitive adsorption on the active cites to ethanol. However, at high OH- concentration (>0.1 mol L-1), the currents mainly come from the redox of Ag(II)/Ag(I) couple, because of the competitive adsorption on the active cites caused by OH-. To obtain high oxidation current at lower oxidation potential, 0.1 mol L-1 NaOH was chosen as an optimum supporting electrolyte.
In order to reveal the electrocatalytic activity of modified carbon paste electrode toward the oxidation of ethanol, the voltammetric experiments were carried out on both modified and unmodified CPEs in the presence of ethanol. Fig. 2 illustrates the CVs of ethanol at the bare carbon paste and SC-MCP/ SB-MCP electrodes in 0.1 mol L-1 NaOH solution at 50mV/s in a potential range of 0.17 to 0.85V vs. Ag/AgCl.
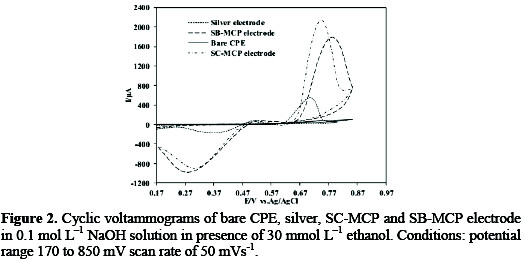
As shown, no oxidation response of ethanol can be seen in the potential range from 0.17 to 0.85V on unmodified electrode, indicating the nonelectroactivity of ethanol on this substrate. From Fig. 2, it can be seen that at the SC-MCP/ SB-MCP electrodes, the anodic currents of ethanol oxidation have been greatly enhanced indicating that the anodic oxidation of ethanol could be catalyzed at silver halide modified carbon paste electrodes. This proves that the silver halide bear the main role in electrocatalytic oxidation of ethanol. In comparison with the data at the bare carbon paste electrode, an increase of 15.1 and 7.6 folds in peak currents (at Ep) of ethanol were observed at the SC-MCP/ SB-MCP electrodes respectively. It can be seen that the electrocatalytic efficiency of SC-MCP electrode is higher than the SB-MCP electrode. Fig. 2 also compares the cyclic voltammogram of SC-MCP/ SB-MCP electrode with that obtained by silver electrode. In comparison with the data at silver electrode, an increase of 12.2 and 6.1 folds in peak current of ethanol was observed at the SCMCP/ SB-MCP electrode respectively.
The following mechanism is proposed for the oxidation of ethanol at the surface of SC-MCP electrode:
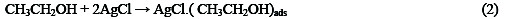
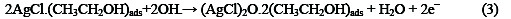
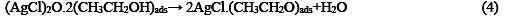
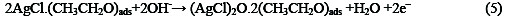
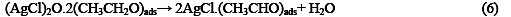
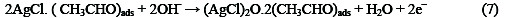
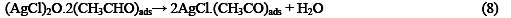
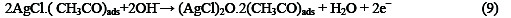
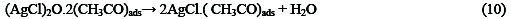
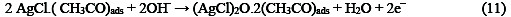
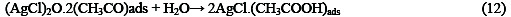
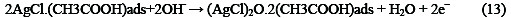
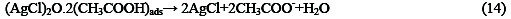
Under the optimal conditions, the relationship between oxidation peak current (ipa/μA) and concentration of ethanol was examined by CV. With the increase of ethanol concentration, the anodic peak current gradually increased (Fig.3).
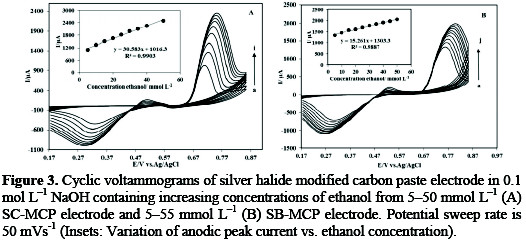
The characteristic shape of cyclic voltammogram in this potential region indicates that the signal is due to the oxidation of ethanol. The voltammetric responses of the modified electrodes towards determination of ethanol have been listed in Table 1.
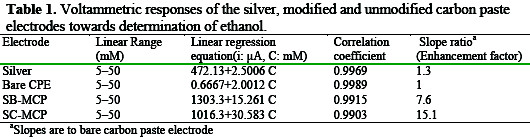
In agreement with the preliminary CV results, it can be seen, from the slopes of the linear regression of the current vs. ethanol concentration plots, that the silver chloride modified carbon paste electrodes display, on a wide ethanol concentration range, a higher electrocatalytic activity with respect to silver bromide modified carbon paste electrode.
In order to provide more evidence, the effect of the scan rate varying from 5 to 100 mVs-1 on the Fig. 4 shows the voltammetric responses of (A) SC-MCP and (B) SB-MCP electrode in a solution containing 30 mmol L-1 ethanol was studied.
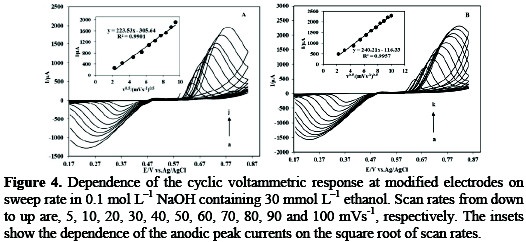
The anodic currents increase and the peak potential shifts as the scan rate increases. The relations between the peak currents obtained from forward CV scan and v1/2 of CV are shown in the insets. The anodic peak currents are linearly proportional to the square root of the scan rates. This behavior suggests that the oxidation process is controlled by diffusion. Also, plots of the scan rate normalized current (Ip/v1/2) vs. scan rate exhibited the characteristic shape of a typical EC' catalytic process. The peak currents for anodic oxidation of ethanol are not proportional to the scan rate. These results indicate that at sufficiently positive potential the reaction is controlled by surface-confined of the ethanol species, which is the ideal case for quantitative applications. Also, it can be seen that the peak potential for the catalytic reduction of ethanol shifts to more positive values by increasing the scan rate, suggesting a kinetic limitation in the reaction between the redox sites of silver halide and ethanol.
Fig. 5 shows the Tafel plots recorded for 20 mmol L-1 ethanol on the (A) bare silver, (B) SB-MCP and (C) SC-MCP electrodes at a scan rate of 10mVs-1.
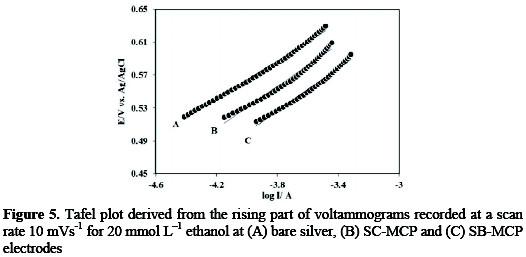
The charge transfer coefficient (α), the number of electrons involved in the rate determining step (nα) and exchange current density (j0) evaluated from Tafel plots have been given in Table 2.
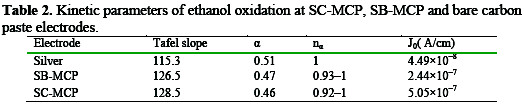
The parameters Tafel slope, α, and nα are very similar for three electrode reported in Table 1, but the j0 for SC-MCP, and SBMCP electrodes are 11.2, and 5.4 folds respectively higher than that of bare silver electrode. The exchange current density for SC-MCP is 5.05×10-7A cm-2, which is 2.1 times higher than that of SB-MCP (2.44×10-7A cm-2).The results indicate that the SC-MCP electrocatalyst is more efficient than the SB-MCP.
Chronoamperometry as well as other electrochemical methods may be used for the investigation of electrode processes at modified electrodes. The chronoamperograms obtained for a series of ethanol solutions with various concentrations as illustrated in Fig. 6A.
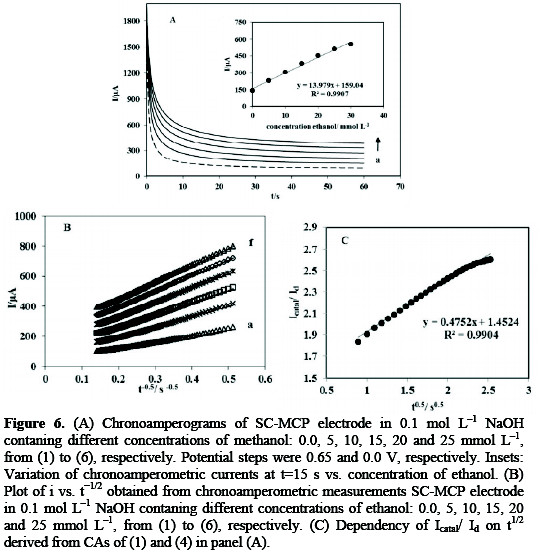
An increase in concentration of ethanol was accompanied by an increase in anodic currents obtained for a potential step of 600mV. The inset of Fig. 6A shows plots of currents sampled at fixed time as a function of ethanol concentration. The response is linearly proportional to the concentration of ethanol in the range of 5-30 mmol L-1. The plot of net current with respect to the mines square roots of time presents a linear dependency (Fig. 6B). This indicates that the transient current must be controlled by a diffusion process. The transient current is due to catalytic oxidation of ethanol, which increases as the ethanol concentration is raised. No significant cathodic current was observed when the electrolysis potential was stepped to 0.00 mV (vs. Ag/AgCl), indicating the irreversible nature of the oxidation of ethanol.
The rate constants of the reactions of ethanol and the ensuing intermediates with the redox sites of the SC-MCP electrode can be derived from the chronoamperograms according to Eq. (1) [20]:
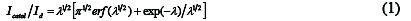
where Icatal is the catalytic current in the presence of ethanol, Id the limiting current in the absence of ethanol and λ=kCt (k, C and t are the catalytic rate constant, bulk concentration of ethanol and the elapsed time, respectively) is the argument of the error function. For λ > 1.5, erf (λ1/2) almost equals unity and Eq. (2) reduces to [20]:

From the slope of the Icatal/Id vs. t1/2 plot (Fig. 6C), the value of k for 15mmol L-1 ethanol was calculated to be 0.4794×104 cm3 mol-1 s-1 . Similar chronoamperograms were collected for SB-MCP electrode. The value of k for 25 mmol L-1 ethanol obtained according to the method described in the above calculated to be 3.7153×104 cm3mol-1s-1 . Since amperometry under stirred conditions has a much higher current sensitivity than cyclic voltammetry, it was used to estimate the lower limit of detection. Figure 7 shows the current-time responses of the SC-MCP electrode to ethanol which was successively added to the electrochemical cell containing 0.1 mol L-1 NaOH under hydrodynamic conditions, while the electrode potential was kept at 0.620.
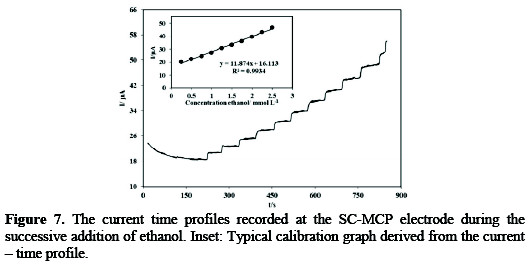
As shown in the figure a well-defined response was observed during the stepwise increasing of ethanol concentration in the range 0.25-2.5 mmol L-1. The linear regression equation of calibration curve is expressed as I (μA) =11.874 CEthanol mmol L-1+16.113 with a correlation coefficient of 0.9934 (n=10). A calibration plot constructed from data of Fig. 7, gives a limit of detection (LOD) and sensitivity 0.31 mmol L-1 and 11.87 μA L mmol L-1, respectively. Similar amperometric response curves were collected for SB-MCP electrode (not shown). The linear regression equation of calibration curve is expressed as I (μA) =0.12 C mmol L-1 μmol L-1 +15.84 with a correlation coefficient of 0.9998 (n=10). The sensitivity and limit of detection (LOD) were found to be 0.12 μA L μmol L-1 and 0.53 μmol L-1, respectively. The reproducibility of the same SC-MCP/ SB-MCP electrodes was examined by measuring the current response to six successive mixed samples containing 30 mmol L-1 ethanol. Relative standard deviation (RSD%) of 2.76% and 2.98% respectively were obtained. To further ascertain the reproducibility of the experimental results, six different SC-MCP/ SB-MCP electrodes were tested towards the oxidation of 30 mmol L-1 ethanol. The peak currents obtained by the six independent electrodes showed RSD of 2.98% for SC-MCP and 3.2% for SB-MCP electrode, confirming that the sensors are reproducible. The stability of the SC-MCP/ SB-MCP electrodes was also tested. After measurements, the modified electrodes were stored at 4°C. The peak current intensity only decreased 6.4% for SC-MCP electrode and 7.6% for SB-MCP electrode for ethanol after one week. The RSD (n = 5) for all these species was less than 9.1% for SC-MCP and 9.9% for SB-MCP electrode. These results above indicated that the modified electrodes possess high selectivity, good reproducibility and well stability.
Conclusions
In the present study, the electrochemical behaviors of ethanol at SC-MCP electrode have been investigated. The modified electrode exhibits excellent and persistent electrocatalytic behavior toward ethanol oxidation compared with the bare carbon paste electrode. The j0 for SC-MCP and SB-MCP electrodes are 11.2 and 5.4 folds respectively higher than that of the bare silver electrode. For SC-MCP electrode, kinetic parameters such as the electron transfer coefficient (α), catalytic reaction rate constant (k) and the number of electrons involved in the rate determining step (nα) for oxidation of ethanol at the SC-MCP surface were calculated as 0.46, 0.4794×104 cm3mol-1 s-1 and 1 respectively.
References
1. Karim-Nezhad G, Dizajdizi B Z, Dorraji P S. Cata Comm. 2011;12:906. [ Links ]
2. Scott K. Taama W M. Argyropoulos P J. J Power Sources. 1999;79:43. [ Links ]
3. Teng Z H, Wang Y J, Wu B, et al. Appl Catal B: Environ. 2008;84:400. [ Links ]
4. Douvartzides S, Tsiakaras P. Solid State Ionics. 2000;849:136. [ Links ]
5. Pang H L, Lu J P, Chen J H, et al. Electrochim Acta. 2009;54:2610. [ Links ]
6. Razmi H, Habibi E S, Heidari H. Electrochim Acta. 2008;53:8178. [ Links ]
7. Salimi A, Abdi K. Talanta 2004;63:475. [ Links ]
8. Kalcher K, Electroanalysis. 1990;2:419. [ Links ]
9. Zare H R, Nasirizadeh N. Int J Electrochem Sci. 2009;4:1691. [ Links ]
10. Ozoemena K I, Staden R S, Nyokong T. Electroanalysis. 2009;21:1651. [ Links ]
11. Shahrokhian S, Amiri M. Solid State Electrochem. 2007;11:1133. [ Links ]
12. Ivanova E V, Sergeeva V S, Oni J, et al. Bioelectrochem. 2003;60:65. [ Links ]
13. Amiri M, Pakdel Z, Bezaatpour A, et al. Bioelectrochem. 2011;81:81. [ Links ]
14. Moscone D, D'ottavi D, Compagnone D, et al. Anal Chem. 2001;73:2529. [ Links ]
15. Wang X L, Wang E B, Lan Y, et al. Electroanalysis. 2002;14:1116. [ Links ]
16. Limoges B R, Stanis R J, Turner J A, et al. Electrochim Acta. 2005;50:1169. [ Links ]
17. Wang X L, Han Z B, Zhang H, et al. Electroanalysis. 2003;15:1460. [ Links ]
18. Jin C, Zhang Z, Chen Z, et al. Electrochim Acta. 2013;87:860. [ Links ]
19. Karim-Nezhad G, Dorraji P S. Electrochim Acta. 2010;55:3414. [ Links ]
20. Karim-Nezhad G, Pashazadeh A, Pashazadeh S. J Korean Chem Soc. 2013;57:322. [ Links ]
*Corresponding author. E-mail address: sa.pashazadeh20@gmail.com
Received 11 August 2014; accepted 25 February 2016

