










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.32 no.3 Coimbra maio 2014
https://doi.org/10.4152/pea.201403213
The Effect of Cathode Materials on Indirect Electrochemical Oxidation of Methyl Orange, Malachite Green and Methylene Blue
Anantha N. S. Rao , Venkatesha T and Venkatarangaiah*
Department of P.G. studies and Research in Chemistry, School of Chemical Sciences, Kuvempu University, Shankaraghatta-577451, Karnataka, India
Abstract
The influence of cathode material on the electrochemical degradation of methyl orange (MO), methylene blue (MB) and malachite green (MG) dyes was investigated. The cathode materials used were platinum (Pt), copper (Cu), zinc (Zn) and aluminum (Al). The electrochemical activity of the selected dyes on the metal cathodes was examined by cyclic voltammetry (CV). The electrochemical treatment was carried out in both divided and undivided cells. The degradation process was monitored by UV-Visible spectroscopy and chemical oxygen demand (COD) measurement. The influence of pH on discoloration and degradation of dyes was studied. The power consumption and current efficiency of the treatment process involving different cathode materials was computed and compared. The role of cathode material in the degradation of dyes has been established.
Keywords: Malachite green; Methylene blue; Methyl orange; Discoloration; Degradation.
Introduction
Dyes are the major constituents of textile industrial effluents. They impart intense color to water. Most of them are highly toxic to aquatic life and also to human beings. The methylene blue (MB), malachite green (MG) and methyl orange (MO) dyes are used on large scale in dying industries (Table 1).
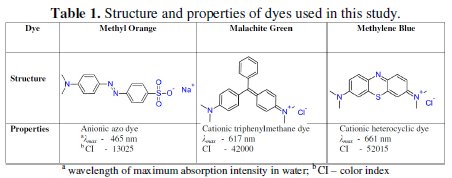
The MG is a cationic water soluble dye used in textile industries for coloring nylon, wool, silk, leather, cotton and jute [1-5]. Also, it finds application as antifungal, antibacterial and anti-parasitical agent in agriculture, fish hatchery and animal husbandry [1,3,5,6]. In anaerobic conditions, the bacteria commonly present in mammalian intestine convert malachite green into toxic colorless leucomalachite green, which poses carcinogenic, mutagenic and teratogenic effects on aquatic life [1, 4, 7]. The degradation products of malachite green are also carcinogenic. Intervention of these species into the food chain is lethal. Another dye, methylene blue, a chlorinated polycyclic synthetic organic compound, has structural similarity with rhodamine b, naphthol blue black and acid orange 7 [8]. MB is a widely used dye in dying cotton, wool, acrylic and silk [9, 10, 11]. It is highly stable and antibiodegradable [11]. Its consumption causes nausea, diarrhea, burning of eyes, vomiting, breathing difficulty, mental disorder, sweating [9, 10]. Methyl orange represents the azo dye family and is commonly used in the titrations of mid strength acids. MO has not shown any harmful effects on rats when used in limited oral and injection range. However, a high acute oral toxicity was noticed [12].
Various methods are used to treat the wastewater containing organic dyes. Physical (precipitation, adsorption) [1, 5, 13-16] chemical [10]; biological [2, 6, 17, 18], photocatalytic [19, 20, 21, 22, 23]; electro-oxidation [4, 9, 24-26]; electro-reduction [27, 28, 29]; photoelectrochemical [30, 31, 32]; electro-Fenton [33, 34, 35] are amongst the most recognized methods.
The electrochemical oxidation method has attracted a large group of researchers in the past three decades. In this method, the efficiency of decontamination of wastewater is dependent on the nature of the electrode. The incineration of organics into CO2 and H2O is possible only by the oxidation reaction. The electrochemical oxidation process is therefore widely appreciated and extensive research on the fabrication of anode material with excellent catalytic activity for the oxidation of organics (direct oxidation) and generation of oxidants (indirect oxidation) is underway.
Nevertheless, the cathode material and reaction at cathode also influence the oxidation of organics. The cathode material can indirectly aid the organic removal by:
• electro-reduction of dissolved oxygen into H2O2, which is an oxidizing agent;
• direct electro-reduction of organics into compounds susceptible to oxidation;
• generating coagulants for the adsorption of organics;
• direct adsorption of organics (porous carbonaceous cathodes).
The halogenated hydrocarbons like, CHCl3, chlorofluorocarbons (CFCs), chlorobenzene, chlorophenols, hexachlorocyclohexane are effectively dehalogenated by electrochemical reduction [33]. The dyes with -SO3-, COO-, -SO2NH2, -OH, hydrophilic groups, and azo linkages are susceptible to electroreduction [36]. Uniform reduction of organics takes place followed by adsorption of intermediates on ACF cathode [36-38]. These reactions taking place in the vicinity of the cathode should be co-operative with the anodic reactions in order to achieve efficient organic removal. It has been reported that the degradation of 2-chlorophenol and 2,4-dichlorophenol using carbon/polytetrafluoroethylene (C/PTFE) O2-fed as cathode and Ti/IrO2/RuO2 as anode was cooperative oxidation. The degradation was achieved by direct and indirect electrochemical oxidation by H2O2 and HO• produced by oxygen reduction at the cathode [39].
Ag, Al, Au, Cu, Ni, Pb, Pd, Pt, Ti, Zn, graphite, glassy carbon, activated carbon fiber (ACF) have been used as cathode materials in the electrochemical treatment of wastewater containing different organic dyes [36-38]. However, only few research teams have tried to analyze the effect of cathode material on the organic removal efficiency [40]. Studies on the co-operation between electrochemical oxidation and reduction reactions in the degradation of aqueous dyes are rarely found in the literature. Also, the contribution of cathode material to the degradation of organics has not been studied. In this paper, we investigate the effect of cathode material and cathodic reactions using different metal cathodes by evaluating the changes in color and COD of the aqueous synthetic dye solutions.
Materials and chemicals
Chemicals
Commercially available malachite green (MG), methylene blue (MB) and methyl orange (MO) dyes (S. D. Fine Chemicals Ltd., India) were used for the studies as received. Millipore water (specific resistance > 18 MΩ at 25 °C, Millipore Elix 3 water purification system, France) was used to prepare the dye solutions and NaCl was used as supporting electrolyte [analytical grade NaCl (0.2% (w/v), 2 g L-1) and 50 mg L-1 dye]. The Zn, Cu, Al metal foils (99.99%) purchased from Sisco research laboratories, Mumbai, India, and Pt foils supplied by Systronics India Ltd. Bangalore, India, were used. The dil. HNO3 (10%), dil. HCl (10%) and NaOH (40%) (HiMedia Laboratories Pvt. Ltd, Bangalore, India) were used for the pretreatment of electrodes. The dialysis membrane-70 with relative pore size of 0.1 μm (HIMEDIA) was used in the divided cell experiments.
Experimental
The electrolysis experiments were performed under galvanostatic condition and the current was drawn from a DC power supply (model PS 618 potentiostat/galvanostat 302/2 A supplied by Chem link, Mumbai). All experiments were carried out at ambient temperature. The electrolysis of dye solution was carried out in both single compartment and two-compartment cells under the current density of 40 mA cm-2 to know the contributions of cathodic and anodic reactions. A volume of 50 cm3 of dye test solution was used in the undivided cell and in each compartment of the divided cell. Pretreated metal foils (Al, Cu, Zn and Pt) with exposed surface area of 0.5 cm2 were used as cathode and Pt (0.54 cm2) as anode in all the experiments. Prior to the experiments, the Al foil was dipped in 40% NaOH, Cu in 10% HNO3 and Zn in 10% HCl, for 1 min, washed thoroughly with Millipore water, abraded with series of emery papers of grade number 660 and 1200 followed by washing in Millipore water. Pt foil was dipped in 10% HNO3 and sonicated for 1 min and thoroughly washed with Millipore water. The working electrode potential was recorded with respect to the saturated calomel electrode (SCE). The electrolysis was performed for 60 minutes under stirring using magnetic bar (550 rpm) to achieve proper mixing and reproducible mass transport onto the electrode surface. The samples were collected at appropriate time intervals to monitor the color and COD change.
Analysis
Electrochemical measurements
The electrochemical behavior of dyes on the cathode surface was evaluated by CV and the potential for hydrogen evolution on different metal cathodes was determined by linear sweep voltammetry (LSV) in 0.2% NaCl (w/v) aqueous solution. The CV measurements were performed at room temperature with a conventional three electrode system connected to software controlled electrochemical work station (CH Instruments 660C, USA). The working electrode was pretreated metal foil (Al, Cu, Pt, Zn). In all the experiments Pt foil was used as auxillary electrode and SCE was used as reference electrode. CV for both blank solution [0.2% NaCl (w/v)] and dye solution (50 mg L-1 + 0.2% NaCl) were recorded and compared.
Spectroscopic and chemical analysis
To follow the degradation process, small aliquots were taken out at regular intervals, diluted with Millipore water, and UV-Visible absorbance spectrum was recorded (HR 4000 UV-Vis Spectrophotometer, UV-Vis-NIR light source, DTMINI- 2-GS, Jaz detector, SP-2102 UVPC (path length = 1 cm)). The percentage color removal with reference to the absorption at λmax was calculated using eq. (1)
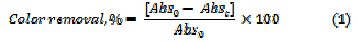
where, Abs0 and Abst are the absorbance at λmax, at time 0 and t minutes of electrolysis, respectively. The mineralization of dye was monitored by the changes in COD (gO2 L-1) as estimated by the open reflux method. The percentage COD removal was calculated using relation (2)
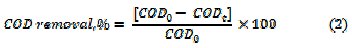
where, COD0 and CODt are the COD at time 0 and t minutes of electrolysis, respectively. The percentage average current efficiency (ACE) for the removal of COD was evaluated by relation (3)
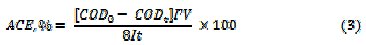
where F is the Faraday's constant (96,485 C moL-1), COD in (gO2 L-1), I is the current (A), V is the volume (L) of electrolyte, t - time (s), 8 - gram equivalent mass of oxygen (g equiv-1). The specific energy consumption per kg COD removal was calculated by eq. (4)
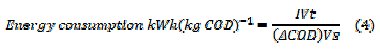
where I is the average applied current (A), V is the average cell voltage (V), t is the electrolysis time (h), Vs is the solution volume (L), ΔCOD is the decay in COD (g L-1).
Results and discussion
Electrochemical studies
The LSV was performed on each cathode material in aqueous NaCl 0.2 % (w/v) solution (Fig. 1) to know the potential for hydrogen evolution reaction (Table 2).
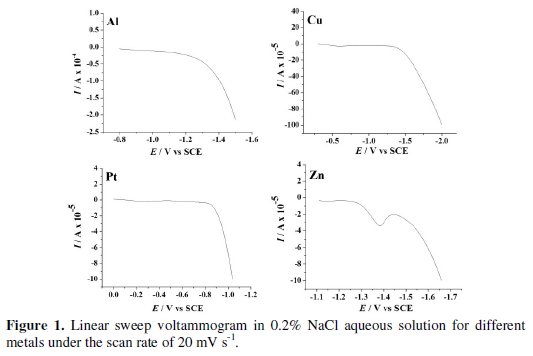
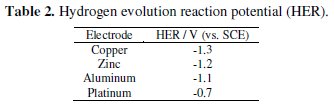
Evidently, Cu exhibited the least activity for hydrogen evolution reaction, whereas on Pt, hydrogen evolution commenced at -0.7 V. Further, the CV was recorded for each dye (50 mg L-1) in 0.2% (w/v) NaCl solution using different cathode materials as working electrode. The operating potential window was selected for each cathode and CV scans were performed (Fig. 2).
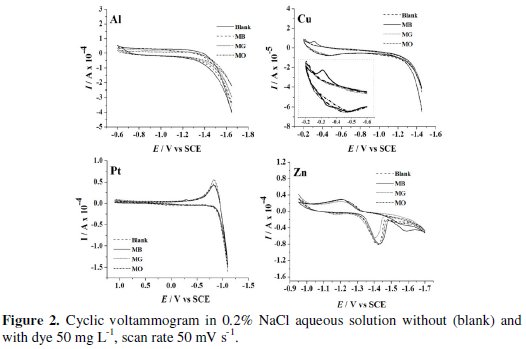
Both MO and MG were found to be electrochemically inactive on all the cathode materials within the operating potential window; whereas peaks corresponding to oxidation and reduction of MB on Cu electrodes were observed in the potential range of -0.2 to -0.4 V vs. SCE (inset).
A peak at -1.42 V vs. SCE was observed in the CV obtained in blank solution on Zn electrode. This peak is due to the electro-reduction of Zn2+ ions liberated from the surface of Zn electrode dipped in NaCl solution. The chemical attack of NaCl on Zn electrode generates Zn2+ ions. These Zn2+ ions get reduced to Zn at -1.42 V vs. SCE before hydrogen evolution. To elucidate the presumption of Zn2+ ion reduction to Zn, the CV scans were performed in 1.0 mM ZnSO4 solution with 0.2 % NaCl as supporting electrolyte under similar conditions. A peak at -1.41 V vs. SCE (Fig. 3) confirms the Zn2+ reduction.
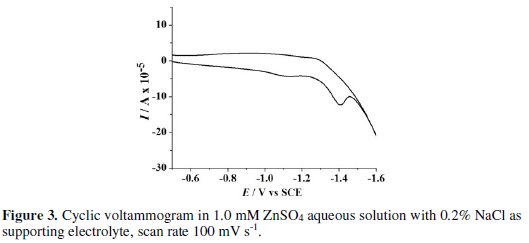
Further, to make sure that this peak is not due to oxygen reduction, a cyclic voltammogram was recorded on Zn electrode in aqueous 0.2 % NaCl solution purged with nitrogen for 30 minutes. The peak at -1.4 V vs. SCE again appeared in CV, negating the chances of O2 reduction reaction. Consequently, the formation of H2O2 by the reduction of O2 is ruled out in the present case.
Electrolysis in undivided cell
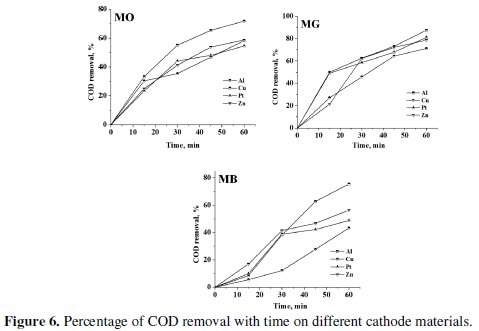
Electrolysis of 0.2% (w/v) aqueous NaCl solution containing 50 mg L-1 dye was performed for a period of 60 minutes under galvanostatic conditions (40 mA cm-2). The progress in degradation was monitored by evaluating the changes in color (Fig. 4 and 5) and COD (Fig. 6) at regular intervals of time.
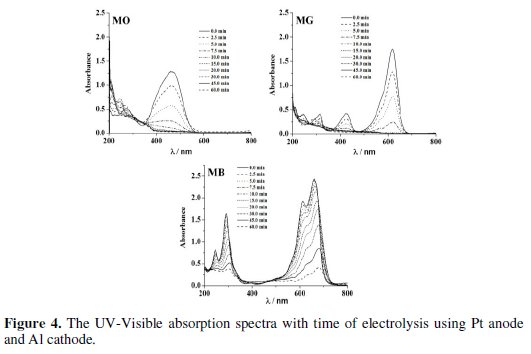
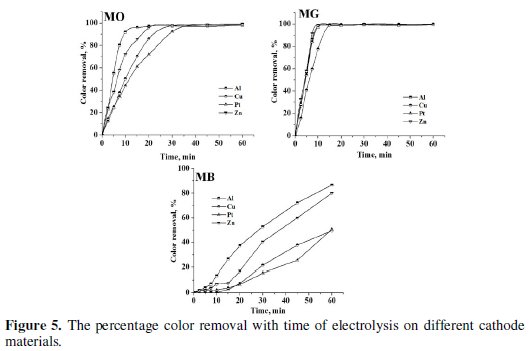
The peaks at 465 nm and 617 nm in the UV-Visible absorption spectrum of MO and MG slowly disappeared with time on electrolysis and complete discoloration was achieved for both MO and MG dyes. However, the rate of discoloration of MO was less as compared to that of MG. 98 % discoloration of MO was achieved within 35-40 minutes of electrolysis, whereas ∼100 % discoloration of MG was attained within 20 minutes. The absorption in the range of 200-300 nm in the UV-Visible spectra of MO dye remained as such even after 60 minutes of electrolysis. This can be ascribed to the aromatic compounds reluctant to oxidation prevailing in the solution. On the other hand, the MB dye was resistant to discoloration. The highest % color removal of MB attained after 60 minutes of electrolysis was only 87 % with Al cathode.
The trend in the COD removal was similar to that of discoloration. The percentage color and COD removal achieved after a period of 60 minutes electrolysis has been tabulated in Table 3.
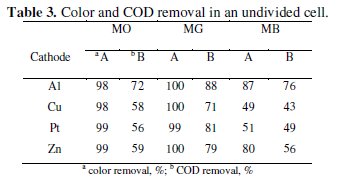
The COD removal of MG was faster and higher as compared to the degradation of MO and MB. The average COD removal of MB was the least amongst the three dyes with Al cathode. The percentage COD removal achieved after 60 minutes of electrolysis under similar conditions followed the order: MG > MO > MB. This suggests that not only the operating conditions, but also the nature of the organic molecule itself influences the degradation process. The heterocyclic aromatic structure of MB was most resistant to degradation and triphenyl methane structure of MG was most vulnerable to electrochemical oxidation.
Though, the trend in the discoloration and degradation was similar with all cathode materials used in the study, the maximum percentage color and COD removal varied with the cathode material. Both discoloration and degradation of all 3 dyes was faster with Al and Zn cathodes. This can be attributed to the fact that Al is chemically attacked by -OH ions generated during the electrolysis [40].
The Al3+ ions thus obtained, form soluble monomeric (Al(OH)y3+) and polymeric (Alx(OH)y3+) species, which later converted into insoluble flocs by the complex precipitation kinetics. These reactions are given in eqs. (5) to (10) [40]:
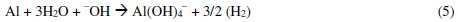
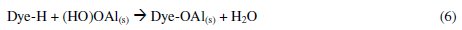
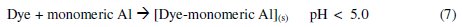
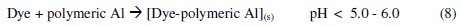

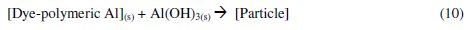
The dye molecule complexes with the Al cationic species and the interaction between them leads to the formation of precipitate. The neutralization of charges leads to the formation of precipitate at pH <5, and adsorption on Al(OH)3 flocs followed by coagulation to form particles occurs at pH >6.5 [40].
The large surface area of Al(OH)3 (s) flocs adsorbs soluble organic compounds [36, 40]. On electrolysis for 60 minutes, the pH of the solution was > 7.8 for all dyes. Under these conditions the reactions in eqs. 9 and 10 are favorable. As a result, the formation of froth and particulates is expected and the same was observed during the electrolysis. Also, the hydrogen evolution and dissolution of cathode material were noticed. The dissolution of Zn metal into Zn2+ ions in NaCl solution is evident by the CV studies, and very low concentration of Zn2+ ions in the test solution is expected.
It is known that at low zinc concentrations, the Zn2+ ion dominates up to pH 8.7, while Zn(OH)2 is the dominant species between pH 8.7 and 11.4. This Zn(OH)2 acts as adsorbent for the adsorption of dye molecules. The following reactions are expected.
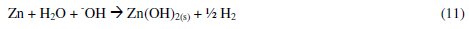

It was established that during electrolysis of aqueous solutions using NaCl, HOCl predominates in the pH range 3-8, and in pH > 8, OCl- is the dominant species (Eqs. 13 to 16) [41, 42].
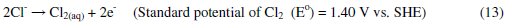
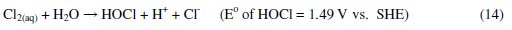
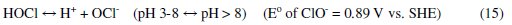
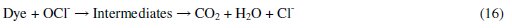
HOCl with higher standard reduction potential is a strong oxidizing agent [40, 43]. In the present case, the pH of dye solution lies in the range where both electro-flocculation and formation of oxidizing agents are favored. The combined effect of oxidation brought about by the active chlorine species HOCl, Cl2, OCl- and the electro-floccuation, resulted in higher COD removal with Al and Zn cathodes.
Degradation in a divided cell
To determine the effect of cathode material and cathodic reactions on the degradation of dyes by indirect electro-oxidation process, experiments were conducted in a divided cell.
The progress in color and COD removal (Fig. 7 - 9) in the anode compartment was monitored and these data are summarized in Table 4.
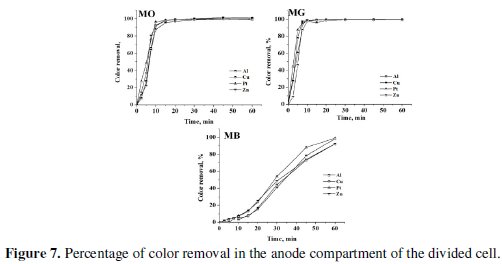
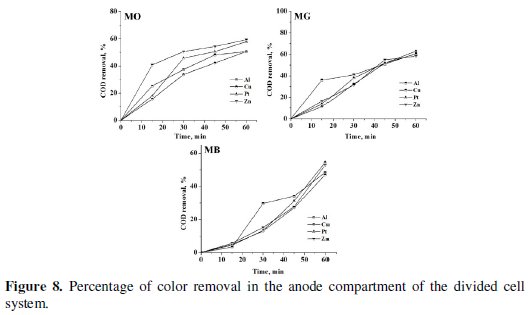
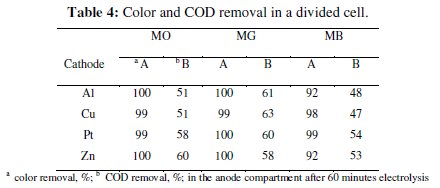
Complete discoloration of both MO and MG was achieved in the anode compartment of the divided cell. For MB, the discoloration was above 90% with all cathodes. The % COD removal was in the order MG > MO > MB. Small % color and COD removal was noticed in the cathode compartment. The color removal observed for MB solution in the cathode compartment was approximately 15% and there was no considerable change in its COD with all 4 cathode materials (< 10%). For MO, the color and COD removal achieved in the cathode compartment was approximately 12% and 8% respectively, irrespective of the cathode material. The % COD removal realized for MG was between 20 and 25 %. These changes in color and COD removal in the cathode compartment of the divided cell can be attributed to the adsorption and / or direct electro-reduction of dyes on the electrode surface.
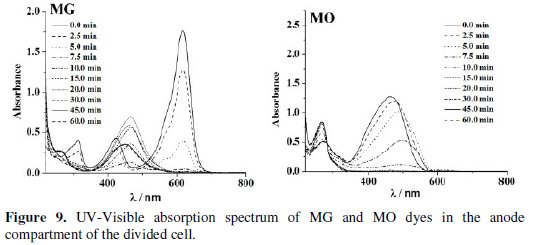
The changes in the UV-Visible spectrum of MB dye during the electrolysis were similar in both undivided and anode compartment of divided cell. However for MO and MG, the UV-Visible absorption spectra exhibited significant differences in divided and undivided cell systems. Three characteristic peaks in the UV- Visible spectrum of MG gradually diminished on electrolysis in the undivided cell, whereas for the solution in the anode compartment of divided cell a new peak around 469 nm was observed. The green solution of MG dye turned yellow after 15 minutes of electrolysis and became colorless at 60 minutes of electrolysis. The MO dye showed a peak at 465 nm for both solutions of undivided and anode compartment of divided cell. During electrolysis, the solution of anode compartment of divided cell exhibited red shift to 500 nm at the beginning of electrolysis and its intensity decreased to zero by the end of the experiment. In the undivided cell, no such shift was noticed but the intensity of the peak gradually reduced to zero at 465 nm.
One of the important observations made is that the pH of the test solution of electrolysis changes during the passage of time in all experiments. In the case of the undivided cell, the pH of the test solution increased on electrolysis irrespective of the dye solution. However, in the divided cell there was rise in pH in the cathode compartment and fall in pH of anode compartment. The pH changes observed on electrolysis in the divided and undivided cell systems have been given in Table 5.
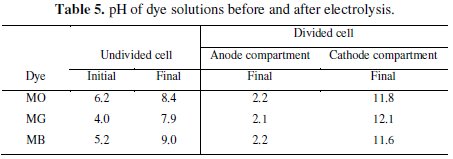
As mentioned earlier, the acidic condition encourages the subsistence of in-situ generated HOCl which oxidized the dyes, resulting in the color and COD removal. As a result, the percentage of discoloration and degradation achieved in the anode compartment was higher than that observed in the undivided cell.
The color of MO and MG dyes are pH sensitive. The MO dye is yellow above pH 4.4 and turns red in pH < 4.4. The pH of as prepared MO solution was 6.2, which increased to 11.8 in the cathode compartment on electrolysis for 60 minutes. In the anode compartment, the MO solution turned red due to reduction in pH which is < 4.4 after electrolysis. The red shift in the UV-Visible absorption at 465 nm is due to this pH change. The discoloration of red solution of MO dye in anode solution was brought about by the in-situ generated active chlorine species. However, the MO dye solution in the cathode compartment did not show any change in the absorption intensity. This is because the pH of cathode compartment turned basic on electrolysis. The MO dye solution is yellow in basic pH and persisted throughout the experiment.
In case of MG, at the end of the experiment, the solution pH of the anode and the cathode compartment were 2.2 and 12.1, respectively. It was noticed that the pH of MG dye solution when varied, imparts green color to the solution in the pH range 1.8 to 11.4. Below pH 1.8 the color of MG dye solution was yellow and this color remains but in pH above 11.8 it is colorless. After the electrolysis, the anode compartment solution pH was 2.2. Therefore, the yellow color of MG was not due to the pH change but due to the intermediate compounds generated by the indirect electro-oxidation of MG. The peak at 256 nm corresponding to the aromatic structure, regularly diminished and reached a minimum. The color removal was due to the oxidative degradation brought about by the active chlorine species. On the contrary, the intense green color of MG solution in the cathode compartment slowly faded and became colorless on electrolysis with time.
This change in color was due to pH change occurred in cathode compartment. The conjugation in the MG structure is lost due to the formation of triphenylcarbinol structure by basic hydrolysis of MG (eq. 17) [40, 44, 45].
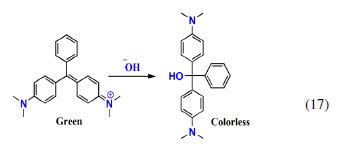
The absorbance observed in the UV-Visible spectrum at 256 nm of the cathode compartment can be attributed to this structure, which persisted even after electrolysis for 60 min (Fig. 10).
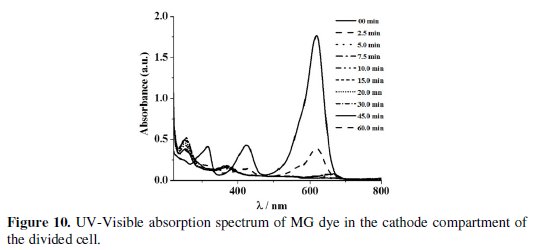
The COD removal for a particular dye solution in the anode compartment of the divided cell was almost the same irrespective of the cathode material used. This is because the degradation was brought about only by the in-situ generated active chlorine species and the concentration of the active chlorine species generated for a particular current density was the same. From the COD removal data given in Tables 3 and 5, the overall degradation achieved in the undivided cell system and in the anode compartment of the divided cell system is comparable.
Energy consumption and efficiency
The specific energy consumption and the average current efficiency for maximum COD removal in 60 minutes of electrolysis was calculated for both undivided and divided systems and presented in Table 6.
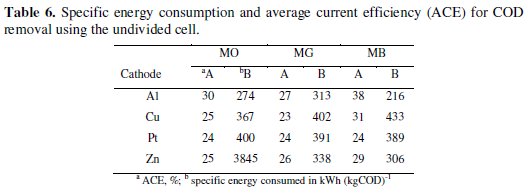
In general, the performance of Al-Pt and Zn-Pt (cathode - anode) pairs was found to be the highest as compared to Cu-Pt and Pt-Pt pairs. As evident from the data, the electrolytic system with Al and Zn cathodes showed the highest efficiency of COD removal with low energy consumption.
The performance of electrolytic cell system with Cu and Pt cathodes was just satisfactory. The MG dye consumed the highest specific energy for the degradation. Although the MG dye color removal was faster than the other two dyes, the degradation of the dye consumed more energy. This can be ascribed to the resistance offered by the intermediates generated by the oxidation of MG. The energy required to reduce the COD to a particular level in the degradation of MG dye was more than that required to reduce the COD to the same level in case of MO and MB.
Conclusion
The degradation of MG, MO and MB was conducted in undivided and divided cell systems with different cathode materials. The in-situ generated active chlorine species brought about the degradation of organics. The performance of Al and Zn metals as cathode materials was found to be higher than that of Cu and Pt as cathodes in this process. The changes in the pH of the wastewater matrix influenced the color and COD removal in divided and undivided cell systems. The basic pH attained on electrolysis and aqueous chloride solution chemically attacked the Al and Zn cathodes, which led to the formation of flocs. This resulted in the higher percentage color and COD removal with Al and Zn cathodes in the undivided cell system. The decoloration and degradation in the anode compartment of the divided cell was comparable with the undivided cell; however, the COD removal achieved for each dye was almost the same with all the cathode materials. The higher acidic pH in the divided cell led to the faster color and COD removal in the anode compartment of the divided cell. The pH variation in the undivided cell was just satisfactory to sustain highly reactive active chlorine species and the formation of flocs, which led to higher color and COD removal with Al and Zn cathodes, whereas Cu and Pt resistant to chemical reaction with -OH and chloride media produced no flocs and hence the showed reduced performance, as is evident from the calculated current efficiency and energy consumption data. Complete discoloration of MG and MO was successfully achieved in all cases. The MB dye showed highest resistance to both decoloration and degradation. This indicates that not only the operating conditions but the cathode material and also the property of dye itself affect the color and COD removal of wastewater containing these dyes.
References
1. Cheng W, Wang SG, Lua L, et al. Biochem Eng Jour. 2008;39:538. [ Links ]
2. Kalyani DC, Telke AA, Surwase SN, et al. Clean Techn Environ Policy. 2012;14:989. [ Links ]
3. Murugesan K, Yang IH, Kim YM, et al. Appl Microbiol Biotechnol. 2009;82:341. [ Links ]
4. Soloman PA, Basha CA, Velan M, et al. Chem Biochem Eng Q. 2010;24:445. [ Links ]
5. Soni A, Tiwari A, Bajpai AK. Res Chem Intermed. 2014;40:913. [ Links ]
6. Daneshvar N, Ayazloo M, Khataee AR, et al. Biores Technol. 2007;98:1176. [ Links ]
7. Henderson AL, Schmitt TC, Heinze TM, et al. App Environ Microbio. 1997;63:4099. [ Links ]
8. Hernandez-Torres ME, Ojeda-Carrera MT, Sanchez-Cantu M, et al. Chemical Papers. 2014;68:1257. [ Links ]
9. El Hajj Hassan MA, El Jamal MM. Port Electrochim Acta. 2012;30:351. [ Links ]
10. Han TH, Khan MM, Kalathil S, et al. Ind Eng Chem Res. 2013;52:8174. [ Links ]
11. Wang Q, Tian S, Ning P. Ind Eng Chem Res. 2014;53:643. [ Links ]
12. Toxicity profile for methyl orange, Bibra toxicology advice and consulting (1992), http://www.bibra-information.co.uk/profile-55.html. Accessed Jan 2014. [ Links ]
13. El-Sharkawy EA, Soliman AY, Al-Amer KM. J Colloid Interf Sci. 2007;310:498. [ Links ]
14. Rajeshkannan R, Rajamohan, Rajasimman M. Front Chem Eng China. 2009;3:146. [ Links ]
15. Ru J, Hua-yue Z, Guang-ming Z, et al. J Cent South Univ Technol. 2010;17:1223. [ Links ]
16. Vecitis CD, Gao G, Liu H. J Phys Chem C. 2011;115:3621. [ Links ]
17. Deng D, Guo J, Zeng G, et al. Int Biodeter Biodegrad. 2008;62:263. [ Links ]
18. Noraini CHC, Morad N, Norli I, et al. Water Air Soil Pollut. 2012;223:5131. [ Links ]
19. Ameen S, Akhtar MS, Kim YS, et al. Colloid Polym Sci. 2010;288:1633. [ Links ]
20. Juan Y, Jun D, Cai ZJ, et al. Chin Sci Bull, Phys Chem. 2010;55:131. [ Links ]
21. Lei J, Li X, Li W, et al. J Solid State Electrochem. 2012;16:625. [ Links ]
22. Shen J, Wu YN, Fu L, et al. J Mater Sci. 2014;49:2303. [ Links ]
23. Xu S, Zhu Y, Jiang L, et al. Water Air Soil Pollut. 2010;213:151. [ Links ]
24. Silva MR, Antonia LHD, Scalvi LVA, et al. J Solid State Electrochem. 2012;16:3267. [ Links ]
25. Panizza M, Barbucci A, Ricotti R, et al. Sep Purif Technol. 2007;54:382. [ Links ]
26. Singh S, Srivastava VC, Mall ID. J Phys Chem C. 2010;117:15229. [ Links ]
27. Fourcade F, Delawarde M, Guihard L, et al. Water Air Soil Pollut. 2013;224:1385. [ Links ]
28. Liu RH, Sheng GP, Sun M, et al. Appl Microbiol Biotechnol. 2011;89:201. [ Links ]
29. Yahiaoui I, Aissani-Benissad F, Madi K, et al. Ind Eng Chem Res. 2013;52:14743. [ Links ]
30. Valatka E, Kulesius. J App Electrochem. 2007;37:415. [ Links ]
31. Zhao Q, Li X, Wang N, et al. J Nanopart Res. 2009;11:2153. [ Links ]
32. Zhou Z, Zhu L, Li J, et al. J Appl Electrochem. 2009;39:1745. [ Links ]
33. Brillas E, Cabot P, Casado J. Electrochemical Methods for Degradation of Organic Pollutants in Aqueous Media. In: Matthew AT (ed). Chemical degradation methods for wastes and pollutants, Environmental and industrial applications. New York, USA;2013. pp 210-273.
34. Minero C, Lucchiari M, Evione D, et al. Environ Sci Technol. 2005;39:8936. [ Links ]
35. Yang S, He H, Wu D, et al. Ind Eng Chem Res. 2009;48:9915. [ Links ]
36. Zhemin S, Wenhua W, Jinping J, et al. J Hazar Mater B. 2001;84:107. [ Links ]
37. Li F, Yanwei Z, Weishen Y, et al. J Hazar Mater B. 2006;137:1182. [ Links ]
38. Li F, Yanwei Z, Weishen Y, et al. Dyes and Pigments. 2008;76:440. [ Links ]
39. Wang H, Wang JL. J Hazar Mater 2008;154:44. [ Links ]
40. MartÃnez-Huitle CA, Brillas E. Appl Catal B Environ. 2009;87:105.
41. Fornazari ALT, Malpass GRP, Miwa DW, et al. Water, Air, Soil Pollut. 2012;223:4895. [ Links ]
42. Malpass GRP, Miwa DW, Santos RL, et al. Environ Chem Lett. 2012;10:177. [ Links ]
43. Song S, Fan J, He Z, et al. Electrochim Acta. 2010;55:3606. [ Links ]
44. Samiey B, Toosi AR. Bull Korean Chem Soc. 2009;30:2051. [ Links ]
45. Soriyan O, Owoyomi O, Ogunniyi A. Acta Chim Slov. 2008;55:613. [ Links ]
Acknowledgements
Authors thank the University Grants Commission, New Delhi (Ref: F. No. 41-231/2012 (SR) Dated 16/07/2012) Govt. of India for providing financial assistance to carry out this work, and Department of chemistry, Kuvempu University for providing laboratory facilities.
*Corresponding author. E-mail address: drtvvenkatesha@yahoo.co.uk
Received 7 May 2014; accepted 23 June 2014

