










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.32 no.2 Coimbra mar. 2014
https://doi.org/10.4152/pea.201402077
Quantum Chemical Studies and Corrosion Inhibitive Properties of Mild Steel by Some Pyridine Derivatives in 1 N HCl Solution
H. Elmsellema , N. Basbasa , A. Chetouania , A. Aounitib , S. Radia,c , M. Messalid and B. Hammoutia,d,*
a Laboratory of Applied Chemistry and Environment (LCAE-URAC18), Faculté des Sciences, Université Mohammed Premier, B.P. 717, M-60000 Oujda, Morocco
b Laboratoire de chimie physique, Centre Régionale des Métiés de l'Education et de Formation CRMEF, Région de l'Orientale, M-60000 Oujda, Morocco
c Centre de l'Oriental des Sciences et Technologies de l'Eau (COSTE), Université Mohamed I, Oujda, Morocco
d Chemistry Department, Faculty of Science, Taibah University, 30002 Al-Madina Al-Mounawara, Saudi Arabia
Abstract
The influence of 2,6-bis-(hydroxy)-pyridine (P1), 2,6-bis-(chloro)-pyridine (P2) and diethyl 1,1'-(pyridine-2,6-diyl)bis(5-methyl-1H-pyrazol2-3-carboxylate ( P3) on the corrosion of steel in 1 N HCl solution has been studied by weight loss measurements, potentiodynamic and impedance spectroscopy methods. The inhibiting action increases with the concentration of these compounds to attain 91,5% at 10-3 M for P3. We note good agreement between gravimetric and electrochemical methods. The polarisation measurements show also that the pyridines act essentially as mixed inhibitors and the cathodic curves indicate that the reduction of proton at the steel surface happens with an activating mechanism. The temperature effect on the corrosion behaviour of iron in 1 M HCl without and with these inhibitors at different concentrations was studied in the temperature range from 313 to 353 K, and allows deducing the apparent activation energy, enthalpy and entropy of the dissolution process and the free energy were determined and discussed. The inhibitors were adsorbed on the iron surface according to the Langmuir adsorption isotherm model at different temperatures and some thermodynamic data for the adsorption process are calculated. The experimental study has been finished by quantum theoretical study; the quantum chemical calculations, based on DFT methods at B3LYP/6-31G** level of theory, were performed, by means of the GAUSSIAN 03 set of programs. Structural parameters, such as the frontier molecular orbital energies (EHOMO and ELUMO), gap of energy ΔE, charge distribution, absolute hardness η and softens σ, fraction of electrons ΔN transferred from pyridine molecules to steel, as well as electronic parameters such as Mulliken atomic populations have been determined. The objective of this quantum theoretical treatment is to attempt to find relationships between their molecular and electronic structures and inhibition efficiency.
Keywords: Pyridine, Inhibition, Corrosion, Steel, Acid, quantum chemical, DFT methods, B3LYP/6-31G base, GAUSSIAN 03.
Introduction
Chemical acid cleaning, acid descaling and pickling processes are widely used in industrial processes to remove mill corrosion scales (oxide scales) from metallic surface in high concentrated HCl media at elevated temperature up to 60 °C. Corrosion never stops but its scope and severity can be lessened. The cost of corrosion has been reported from many studies as to be of the order of 1% to 5% of GNP for any country. In a sense, the use of inhibitors is one of the best known methods of corrosion protection. The addition of the inhibitor is necessary to avoid the attack on the metal and should be effective even under severe conditions in concentrated acid. The selection of a suitable inhibitor depends on its chemical structure, concentration, nature of the metal and on the type of acid. The selection of this inhibitor is based also on the fact that it contains a ring as reactive centres through which it can be adsorbed readily on the metal surface. It is readily soluble in the medium and it does not cause any health hazards, hence its use as corrosion inhibitor is safe. So, the inhibition efficiency of organic compounds depends on the structure of the inhibitor, the characteristics of the environment in which it acts and the mode of interaction with the metal surface. Organic compounds containing electronegative functional groups and p-electron in triple or conjugated double bonds are usually goods inhibitors. It was shown that the protective properties of such compound depend upon their ability to reduce the corrosion rate and are enhanced at higher electron densities around the oxygen and nitrogen atom, which are the major adsorption centres [1, 2]. In acid environment, the inhibitor acts generally by chemisorption and/or physical adsorption at the metal surface. To be effective, an inhibitor must also displace water molecules from the metal's surface and interact with anodic and cathodic reaction sites to retard the oxidation and/or reduction corrosion reaction.
It is widely accepted that organic inhibitors exhibit corrosion via adsorption. The inhibition mechanism is a separation process involving (i) the inhibitor is adsorbed on the surface of the metal forming a compact protective thin layer and (ii) the inhibitor forms a precipitate on the surface of the metal, acting on the aggressive media to form protective precipitates or remove the aggressive agents [20]. Adsorption can also be described by two main types of interaction, which are physisorption and chemisorption, where [21, 22]:
Physisorption: involves electrostatic forces between ionic charges or dipoles on the adsorbed species and the electric charge at the metal/solution interface. The heat of adsorption is low, thus, this adsorption is only stable at relatively low temperature.
Chemisorption: involves charge transfer or sharing from the inhibitor molecules to the metal surface to form a coordinate type bond. This type of adsorption is known to have much stronger adsorption energy compared to the other mode of adsorption. Thus, such bond is more stable at higher temperatures.
Adsorption is generally explained by the formation of an adsorptive film of a physical or chemical character on the metal surface. In continuation of our work on development of organic compounds as acid inhibitors, we have synthesised an organic inhibitor namely 2,6-Bis-(hydroxy)-pyridine (P1), 2,6-Bis-(chloro)pyridine (P2) and diethyl 1,1'-(pyridine-2,6-diyl)bis(5-methyl-1H-pyrazol2-3carboxylate (P3), present in Fig. 1.
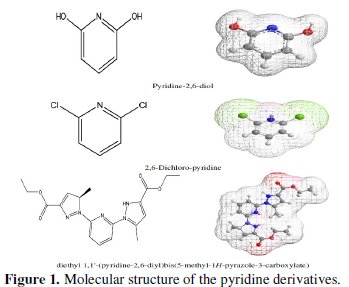
The synthesis of new organic molecules offers various molecular structures containing several heteroatoms and substituents. Weight loss tests and electrochemical techniques have been used to study the effect of addition of this compound on the corrosion of steel in HCl acid solution with a view to study it's inhibiting properties on corrosion of steel in 1 N HCl in the 298-353 K range. Furthermore, quantum chemical calculations have been performed at B3LYP/6-31G(d,p) level to complement the experimental evidence. We aim to investigate whether there is a clear relationship between the experimentally determined inhibition efficiencies of the studied inhibitors. A number of quantum-chemical parameters are directly extracted from the output files (EHOMO, ELUMO, μ), when other parameters need to be computed separately (IP, EA, ΔE, χ, η, σ and ΔN).
Experimental
Inhibitors
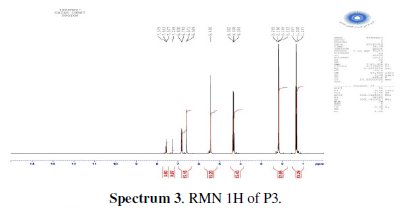
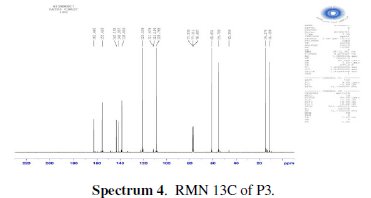
The chlorination of the commercially available P1 carried out in dichloromethane and thionyl chloride gave the compound P2 with a good yield (87 %). One equivalent of the last product P2 was condensed with two equivalents of 3(5)carboxymethyl- 5(3)-methylpyrazole under solid-liquid phase transfer catalysis to favour the a-isomer. Thus, one isolated major product P3 in 72% as the aaisomer was formed. The synthesis of target products was illustrated in scheme 1 and the characterization is presented in spectra 1, 2 and 3, 4 respectively for P2 and P3.
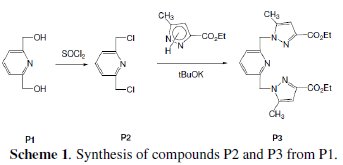
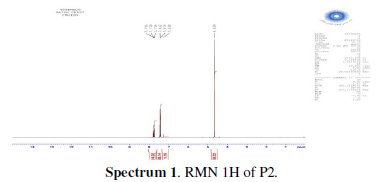
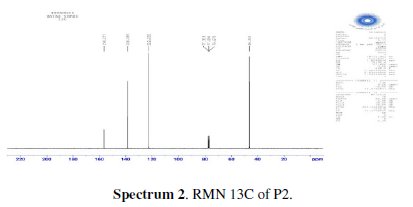
The analytical and spectroscopic data are conforming to the structures of the compounds formed. For 2,6-bis(chloromethyl)pyridine (P2) (Fig. 1): Yield 87%. 1H RMN (300MHz, CDCl3) δ ppm: 4.65 (s, 4H, Cl-CH2-), 7.42 (d, 2H, Hmeta), 7.76 (d, 2H, Hpara). 13C RMN (75 MHz, CDCl3) δ ppm: 46.24 (Cl-CH2-), 122.23 (Cortho), 138.38 (Cmeta), 156.25 (Cpara). Anal. Calcd. for C7H7Cl2N: C 47.76, H 4.01, N 7.96, Found: C 47.79, H 4.08, N 7.88; m/z (M+): 175.
For ethyl1-((6-((3-(ethoxycarbonyl)-5-methyl-1H-pyrazol-1-yl)methyl)pyridin-2yl) methyl)-5-methyl-1H-pyrazole-3-carboxylate (P3) (Fig. 1): Yield 72%. 1H RMN (300MHz, CDCl3) δ ppm: 1.31 (t, 6H, CH2-CH3), 2.16 (s, 6H, pyrazolCH3), 4.32 (q, 4H , CH2-CH3), 5.43 (s, 4H, N-CH2-), 6.56 (s, 2H, pyrazol, CH), 6,80 (d, 2H, Hmeta), 7.53 (d, 2H, Hpara). 13C RMN (75 MHz, CDCl3) δ ppm: 11.18 (Et), 11.27 (Et), 54.78 (N-CH2), 60.85 (CH2-Py), 108.76 (Py), 120.53 (Py), 138.66 (Py), 140.88 (Bz), 143.15 (Bz), 162.44 (Bz). Anal. Calcd. for C21H25N5O4: C 61.30, H 6.12, N 17.02, Found: C 61.29, H 6.15, N 7.07; m/z (M+): 411.4.
Gravimetric, polarisation and impedance spectroscopy measurements
Prior to all measurements, the steel samples (0.09 % P; 0.38 % Si; 0.01 % Al; 0.05 % Mn; 0.21 % C; 0.05 % S and the remainder iron) were ground with different emery papers up to 1000 grade, washed thoroughly with bidistilled water, degreased and dried with acetone.
The aggressive solution (1 N HCl) was prepared by dilution of analytical grade 37% HCl with bidistilled water.
Gravimetric measurements were carried out in a double walled glass cell equipped with a thermostat-cooling condenser. The solution volume was 100 cm 3. The steel specimens used had a rectangular form (2 cm × 2 cm × 0.05 cm). The electrolysis cell was a cylinder of Pyrex closed by a cap containing five openings. Three of them were used for the electrodes. The working electrode was mild steel with the surface area of 1 cm2. Before each experiment, the electrode was polished using emery paper until 1200 grade. After this, the electrode was cleaned ultrasonically with distillate water. A saturated calomel electrode (SCE) was used as a reference electrode. All potentials were given with reference to this electrode. The counter electrode was a platinum plate of surface area of 1 cm2. The temperature was thermostatically controlled at 298 ± 1 K. The working electrode was immersed in the test solution during 30 minutes until a steady state open circuit potential (Eocp) was obtained. The polarization curve was recorded by polarization from -800 mV to 100 mV under potentiodynamic conditions corresponding to 0.5 mV/s (sweep rate) and under air atmosphere. The potentiodynamic measurements were carried out using a VoltaLab100 electrochemical analyser, which was controlled by a personal computer. AC- impedance studies also were carried out in a three electrode cell assembly. The data were analysed using Voltamaster 4.0 software. The electrochemical impedance spectra (EIS) were acquired in the frequency range 10 kHz to 1 NHz at the free corrosion potential. The charge transfer resistance (Rct) and double layer capacitance (Cdl) were determined from Nyquist plots.
Quantum chemical calculations
Computational chemistry (also known as molecular modelling) is the application of computer-based models to the simulation of chemical processes and the computation of chemical properties. It accounts for roughly a third of the supercomputer usage worldwide. However, improved desktop computers mean that these methods are also becoming more available to scientists who are not purely computation lists. According to a recent textbook, "Today, the situation has been reached where, in many cases, the computational chemist can substitute the computing machine for the test tubeâ. Computational chemistry is a valuable tool for experimental chemists to bypass tedious, time consuming, costly, and sometimes dangerous experiments. In the drug industry, computer design of molecules with specified properties is now becoming more common. Furthermore, computational chemistry allows one to investigate molecules that are too unstable to be studied experimentally, to analyze quantities (such as atomic charges) that are not experimentally observable, and to rectify incorrect experimental assignments. Also, computational chemistry allows one to both calculate certain quantities (such as heats of formation) with more accuracy than can be determined experimentally (special cases only) and improve one's general understanding of chemical phenomena.
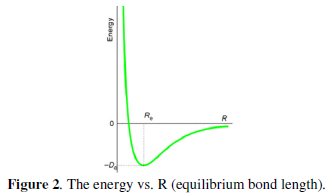
The process of finding the arrangement of nuclei for which the potential energy is a minimum is known as geometry optimization. The potential energy as a function of internuclear distance for a diatomic molecule is shown in the diagram below. Note that it takes a lot of energy to get atoms very close to each other; there is a distance with minimum energy Ro, and it takes energy to pull the atoms apart farther than Ro. Why does the energy approach zero as the intermolecular distance gets very large? The lowest-energy is at Ro (equilibrium bond length), where the derivative of the potential energy curve is zero. The geometry optimization procedure involves making an initial guess for the geometry and then calculating the derivative (gradient) of the potential energy with respect to each of the nuclear coordinates. The gradient represents the force acting upon each atom. These energy gradients are then used to obtain a new geometry that is likely to be closer to the equilibrium geometry. This process is repeated until the energy gradients (forces) approach zero, indicating that equilibrium geometry has been found (Fig. 2).
Recently, quantum chemical calculations have been performed enormously to complement the experimental evidences. Quantum chemical methods and molecular modelling techniques enable the definition of a large number of molecular quantities characterizing the reactivity, shape and binding properties of a complete molecule as well as of molecular fragments and substituents. Density Functional Theory is another type of ab initio calculation that is much less expensive (in terms of CPU time, memory, and disk space) than HF and QMC because it is based on total electron density rather than wave functions. It is now generally agreed that density functional theory provides the most promising approach to accurate quantum chemical calculations for large systems. Ab initio calculations are qualitatively good, and for very small molecules give excellent quantitative results. The recently developed density functional theory (DFT) quantum chemical calculations, with the Beck's three parameter exchange functional along with the Lee-Yang-Parr nonlocal correlation functional (B3LYP) [26-28] with 6-31G* basis set, is implemented in Gaussian 03 program package [29], have been applied widely to compute electronic properties possibly relevant to the inhibition action.
This approach is shown to yield favourable geometries for a wide variety of systems. This basis set gives good geometry optimizations. The geometry structure was optimized under no constraint. Knowing the orientation of the molecule, favourable configurations, atomic charges and steric and electronic effects would be useful for better understanding of the inhibitor performance. It has been stated that the experimental data can be correlated well with quantum chemical parameters such as electron density, ionization potential, total energy, dipole moment, charge density, highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbital energies and the gap between them, the absolute hardness η and softens σ, the fraction of electrons ΔN transferred from pyridine molecules to steel as well as electronic parameters such as Mulliken atomic populations.
Results and discussion
Weight loss tests
The gravimetric method (weight loss) is known to be the most widely used method of monitoring inhibition efficiency. Mild steel specimens of dimension 4 × 3 × 0.1 cm were used in these studies. The specimens were weighed before and after the tests using an analytical balance with a precision of 0.01 mg. The specimens were taken out of immersion, washed, dried and reweighed accurately to determine the weight loss of mild steel. The weight loss measurements were carried out by weighing the prepared specimens before and after immersion for 6 h in 100 mL stagnant test solutions of 1 N HCl in the presence and absence of various concentrations of 2,6-bis-(hydroxy)-pyridine (P1), 2,6-Bis-(chloro)pyridine (P2) and diethyl 1,1'-(pyridine-2,6-diyl)bis(5-methyl-1H-pyrazol2-3-carboxylate (P3) compounds tested at different concentrations.
From the weight loss data, the percentage inhibition efficiency, and maintained in a thermostated water bath, (IEw%), was calculated at different concentrations and at different temperatures by equation 1, where Wcorr and W°corr are the corrosion rates of steel with and without organic compound, respectively.
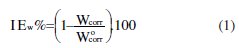
Table 1 regroups the results of weight loss of steel in 1 N HCl with and without the addition of various concentrations of the pyridines compounds.
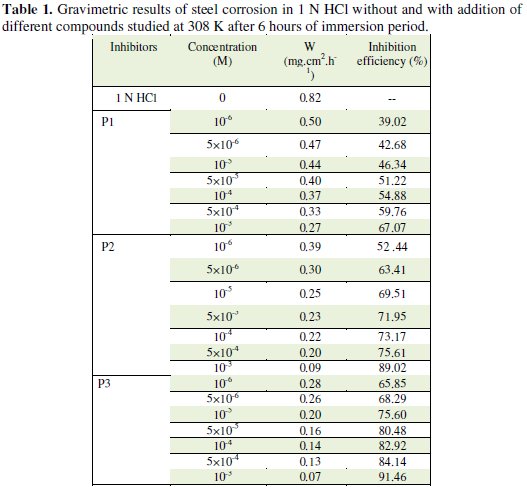
It was found that the value of Ew% increases with an increase in the concentration of additives compounds P1, P2 and P3, which confirmed that the number of molecules adsorbed increased over the steel surface, locking the active sites from acid attack and, thereby, protecting the metal from corrosion. At the highest concentration of 10-3 M of each additive studied, the Ew% attained was 67.07%, 89.02% and 91.46% for P1, P2 and P3, respectively, which confirmed that both of the additives were very effective as inhibitors.
It is interesting to note that the inhibiting effect of P3 can be attributed to its adsorption at the metal solution interface; the adsorption is owing to the presence of more active centres (several nitrogen atoms and a great deal of π-electrons of rings) for adsorption with positively charged steel surface. It has been reported that the nitrogen atoms are the adsorption centres for their interaction with the metal surface and these compounds are usually good inhibitors.
Polarisation results
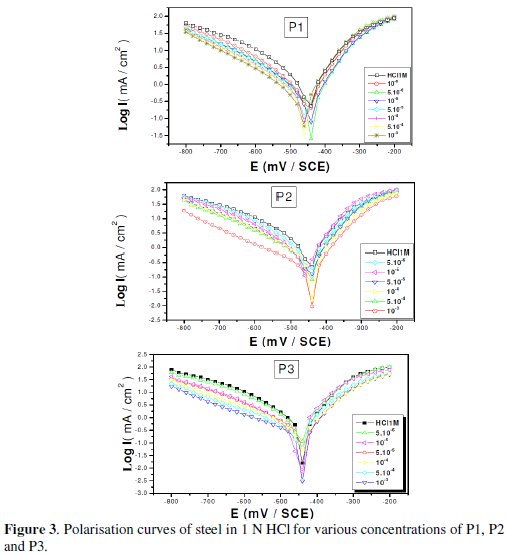
Polarization measurements have been carried out in order to gain knowledge concerning the kinetics of the anodic and cathodic reactions. Fig. 3 shows the influence of 2,6-Bis-(hydroxy)-pyridine (P1), 2,6-Bis-(chloro)-pyridine (P2) and diethyl 1,1'-(pyridine-2,6-diyl)bis(5-methyl-1H-pyrazol2-3-carboxylate (P3) concentration on the cathodic and anodic polarisation curves of steel in 1N HCl.
Evaluation of EI(%) can also be performed through electrochemical experiments which consist of the determination of intensity/potential curves.
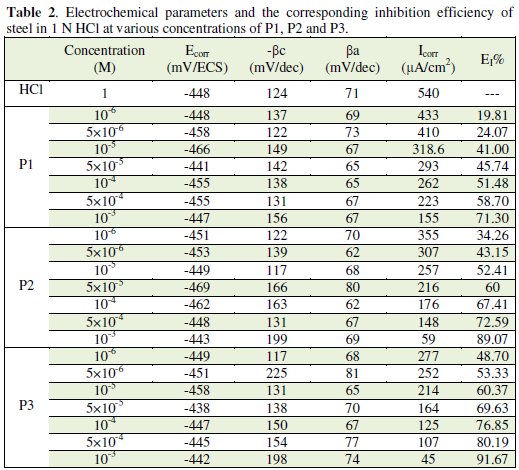
Various corrosion kinetic parameters such as corrosion current (Icorr), corrosion potential (Ecorr), cathodic Tafel slope (βc) derived from the polarization curves, Fig. 3, and inhibition efficiencies (EI%) of P1, P2 and P3 are given in Table 2.
In the case of polarization method, the inhibition efficiency EI(%) is determined by the relation 2, where Icorr and I0corr are the corrosion current density values with and without inhibitor, respectively, determined by extrapolation of the cathodic Tafel lines to corrosion potential.
Under the experimental conditions performed, the cathodic branch represents the hydrogen evolution reaction, while the anodic branch represents the iron dissolution reaction. They are determined by extrapolation of Tafel lines to the respective corrosion potentials. Some authors proposed the following mechanism for the corrosion of iron and steel in acid solution [6,9]:
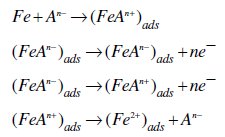
The cathodic hydrogen evolution
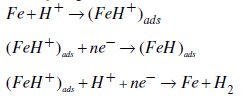
As it is shown in Fig. 3 and Table 2, cathodic polarisation curves rise to parallel Tafel lines indicating that the hydrogen evolution reaction is activation controlled. Thus the presence of pyridine compounds does not affect the mechanism of this process.
The addition of pyridine molecules causes a decrease of the current density. The values of corrosion potential (Ecorr) and cathodic Tafel slope (βc) remain almost constant upon the addition of inhibitor concentration. The results demonstrate that the hydrogen reduction is inhibited and that the inhibition efficiency increases with P3 inhibitor concentration to attain 91,67% at 10-3 M. It seems that after potential corrosion, the presence of the pyridine studied does not change the current versus potential characteristics. Compared to the blank sample, the anodic curves of the carbon steel electrode in the acid solution shift obviously to the direction of current reduction as adding the P1, P2 and P3, which implies that the organic compounds can also suppress the anodic reaction (steel dissolution) and the variation in the anodic Tafel slope Δa may be due to the chloride ions/or inhibitor molecules adsorption onto the carbon steel surface. Based on the marked decrease of the cathodic and anodic current densities upon introducing the inhibitor in the aggressive solution, this inhibitor is considered as a mixed-type inhibitor. This means that the addition of pyridine compounds reduces the anodic dissolution and also retards the cathodic hydrogen evolution reaction.
Electrochemical impedance spectroscopy (EIS)
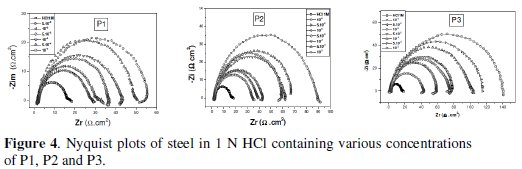
Impedance measurements are being ever more widely used in the fields of electrochemistry and corrosion, although they require costly apparatus and the interpretation is sometimes quite difficult. With impedance measurements it is possible to get information about various electrochemical processes and provide information on both the resistive and capacitive behaviour of the interface, making possible to evaluate the performance of the studied inhibitors. Before each EIS experiment, as done with Tafel experiments, the electrode was allowed to corrode freely for one hour to obtain a steady-state open circuit potential, corresponding to the corrosion potential, Ecorr, of the working electrode. EIS was carried out on a newly polished steel surface in acidic solution in the absence and presence of inhibitor at open circuit potential at 298 K after 30 min of immersion. Nyquist plots of steel in uninhibited and inhibited acid solutions containing various concentrations of pyridine compounds are shown in Fig. 4.
The charge transfer resistance, (Rt), values were calculated from the difference in impedance at lower and higher frequencies. To obtain the double capacitance (Cdl), the frequency at which the imaginary component of the impedance is maximum (-Zmax) was found and Cdl values obtained from equation 3:
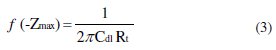
The percentage inhibition efficiency was calculated from the charge-transfer resistance by the following relation 4, where Rt and Rt (inh) are the charge transfer resistance values with and without inhibitor, respectively:
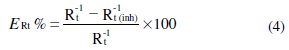
The impedance parameters derived from these investigations are given in Table 3.
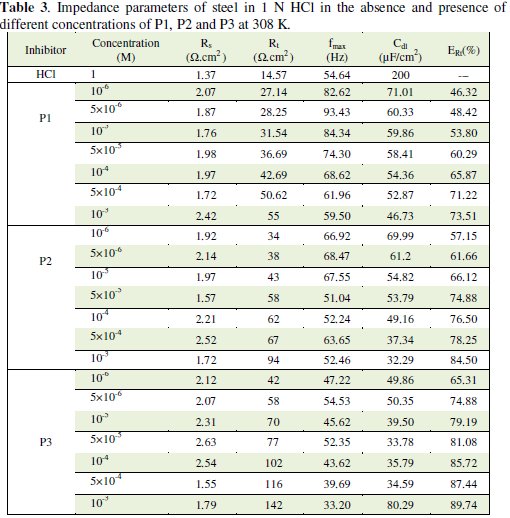
The inhibition efficiency, ERt%, was found to increase with an increase in the concentration of P1, P2 and P3. It is observed that Rt values increase with the increase of the pyridines compounds concentration indicating an insulated adsorption layer formation. This suggests that the role of the inhibitor molecules is preceded by the adsorption at the metal-solution interface. Therefore, P1, P2 and P3 molecules function by adsorption at the metal-solution interface. The adsorption of P1, P2 and P3 on the steel electrode leads to a decrease of its electrical capacity because they might have displaced the water molecules and other ions originally adsorbed on the metal surface. Decreasing the capacitance, Cdl, with increasing concentrations of P1, P2 and P3, resulted from the decrease in local dielectric constant due to the increase of the electrical double layer thickness with the formation of a protective layer on the electrode surface. Effectively and according to the Helmholtz model, the double layer capacitance Cdl is given by equation 5, where δ is the thickness of the deposit, S is the surface area of the electrode, ε0 is the permittivity of the air and ε is the medium dielectric constant:

The decrease in Cdl values (Fig. 4) may be attributed either to a decrease of local dielectric constant [33] or to the thickness of the adsorbed inhibitor layer on the metal surface [34]. The impedance diagram consists of large capacitive loops with small inductive one at low frequency values. In fact, the presence of pyridine compounds enhances the value of Rt in acidic solution indicating a charge transfer process mainly controlling the corrosion of steel. Values of double layer capacitance are also brought down to the maximum extent in the presence of pyridines.
Another evidence for the effective adsorption of P1, P2 and P3 on the steel surface can be given from the observation that the maximum frequency (fmax) of the capacitive loop of the uninhibited solution is decreased with increasing inhibitor concentration. The results obtained from EIS have shown a similar trend to those obtained from electrochemical polarizations and weight loss measurements.
Effect of temperature
The effect of temperature on the inhibited acid-metal reaction is very complex, because many changes occur on the metal surface such as rapid etching, desorption of inhibitor and the inhibitor itself may undergo decomposition. The dependence of the corrosion rate on temperature can be expressed by the Arrhenius equation and transition state equation as presented in Eqs. (6) and (7), respectively.

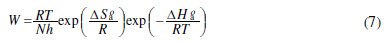
The apparent activation energies can be determined by the relation (6), where E'a and Ea are the apparent activation corrosion energies with and without inhibitors, R is the universal gas constant, T is the absolute temperature and A is the pre-exponential factor.
Other kinetic data (enthalpy and entropy of corrosion process) are accessible using the alternative formulation of Arrhenius (equation (6)) and transition state (equation (7)), where h is Planck's constant, N is Avogadro's number, ΔSa° and ΔHa° are the entropy and enthalpy of activation, respectively.
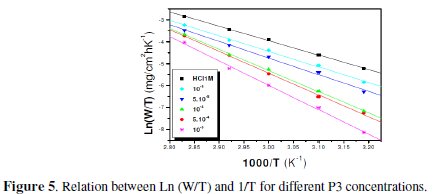
The plots of ln (W/T) vs. the reciprocal of temperature, Fig. 5 and 6, show straight lines with a slope equal to (-ΔHa°/R) and an intercept of (Ln R/Nh + ΔSa /R).
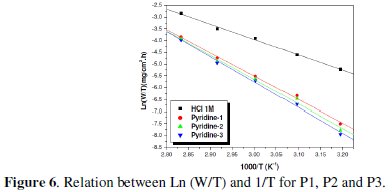
The values of ΔHa and ΔSa are also presented in Table 4 and 5.
The corresponding results are given in Tables 6 and 7.
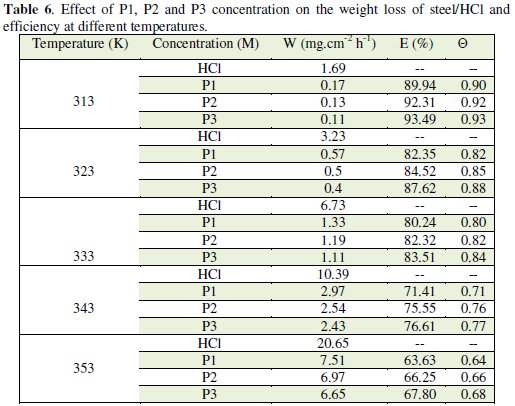
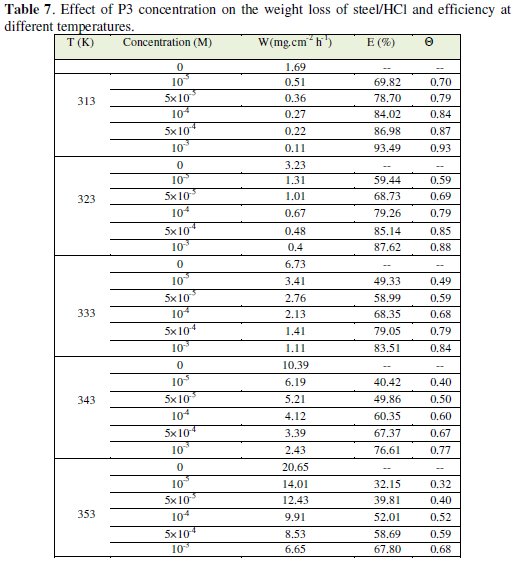
We note that the corrosion rate increases with the rise of temperature both in inhibited and uninhibited acids. It is clear that inhibition corrosion rate increased with increase in temperature due to the acceleration process of corrosion in HCl at elevated temperatures. The inhibition efficiency increased with all inhibitors (P1, P2 and P3) concentration but decreased with the rise of temperature.
This result suggested that the corrosion inhibitions of steel might be caused by the adsorption of P1, P2 and P3 compounds onto the steel surface from acidic solution. As temperatures increased a competition between adsorption and desorption occurs on the steel surface. The increase of corrosion rate is more pronounced with the rise of temperature for blank solution. This can be explained by the decrease of the strength of adsorption processed at elevated temperature and suggested a physical adsorption mode.
The linear regression plots between ln(W) and 1000/T are presented in Fig. 7 and 8.
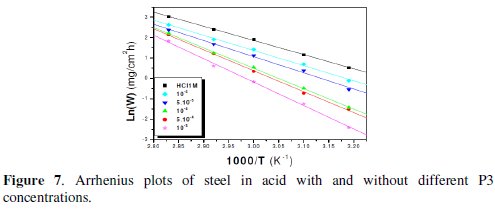
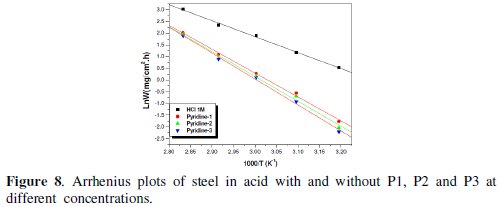
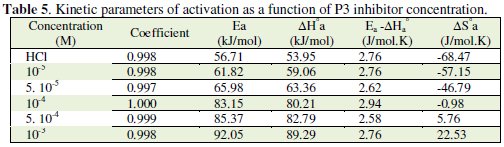
The calculated activation energies, Ea, and pre-exponential factors, A, at different concentrations of the inhibitor are collected in Table 5.
Analysis of the temperature dependence on inhibition efficiency as well as comparison of corrosion activation energy in the absence and presence of inhibitors gives insight knowledge on the possible mechanism of the inhibitor adsorption. Effectively, the change of the values of the apparent activation energies may be explained by the modification of the mechanism of the corrosion process in the presence of adsorbed inhibitor molecules [3-6]. A decrease in inhibition efficiency with rise in temperature with an increase in Ea in presence of the inhibitor compared to the absence is frequently being interpreted as the formation of an adsorptive film of physisorption [34].
This type of inhibitor retards corrosion at ordinary temperatures but inhibition is diminished at elevated temperature. It's observed that the Ea value is increased in the presence of the inhibitor, indicating the poorer performance of the compound at higher temperatures. The addition of inhibitor modified the values of the apparent activation energies; this modification may be attributed to the change in the mechanism of the corrosion process in the presence of the adsorbed inhibitor molecules. The lower value of the activation energy (Table 5), of the process in the inhibitor's presence when compared to that in its absence is attributed to its chemisorption, while the opposite is the case with physical adsorption. This type of inhibitor retards corrosion at ordinary temperatures but inhibition is diminished at elevated temperature.
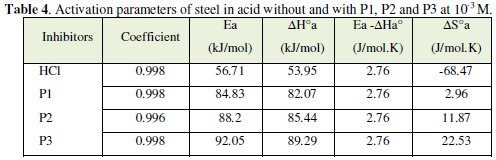
In order to study the data collected in Table 4, the effect of temperature on the corrosion inhibition of mild steel in the acid medium, and to determine the activation energy of the corrosion process, gravimetric measurements have been taken at various temperatures (313-353 K) in the presence and absence of P3 at different temperatures for 1 h. All calculated parameters are given in Table 4. It's observed that the Ea value is increased in the presence of the inhibitor, which indicates the poorer performance of the compound at higher temperatures. This increase of activation energy may be attributed to the process of electrostatic adsorption of P3 on the steel surface by increasing the energy barrier for the corrosion process [4, 7, 8].
The positive values of ΔH reflect the endothermic nature of the mild steel dissolution process. The values of Ea and ΔH* enhance with an increase in the concentration of P3, suggesting that the energy barrier of the corrosion reaction increases as the concentration of P3 is increased. This means that the corrosion reaction will further be pushed to surface sites that are characterized by progressively higher values of Ea as the concentration of the inhibitor becomes greater [40]. Inspection of the kinetic data obtained in Tables 4 and 5, shows that all parameters of corrosion process increase with the inhibitor concentration. The negative values of ΔS imply that the disorderness is increased on going from reactant to product. It is observed that the shift of ΔS to more positive values on increasing the concentration of the extract is the driving force that can overcome the barriers for the adsorption of inhibitor onto the mild steel surface. In the present study, it could be found that the ΔS* increases with the increase in concentration of P3, and passes from the -57.15 at 10-5 M to 22.53 J-1 mol at 10-3 M; furthermore, we notice as the ΔS changes its sign from negative to positive as a result of possible change in the mechanism of the corrosion process and in the disordering taking place on going from reactants to the activated complex. The obtained result was similar to the author's previous outcome [1, 9]. The increase of Ea and ΔHa° with an increase in the P3 concentration suggests that the energy barrier of corrosion reaction increases with the inhibitor's concentration. In this work, P3 contains the main active centres of molecule and cycle ring. The presence of free electron pair in the nitrogen atom favours the adsorption of the inhibitor. It is apparent from the results that the molecular structure of this compound is essential in affecting the corrosion inhibition [10-12]. This observation clearly proves that the inhibition is due to the formation of an insoluble stable film from the process of chemisorption on the metal surface. The formation of a stable film on the metal surface through chemisorption is tested and reported [2, 8, 13]. It is found that an adsorbed film is formed on mild steel surface which is responsible for the inhibition of corrosion in the acid. The protective activity of the adsorbed film in the acid solution is retained for a period of time when specimens, dipped in inhibited acid, are transferred to fresh acid without inhibitor.
Adsorption isotherm
It is useful to describe the adsorption process by an appropriate adsorption isotherm. The extent of corrosion inhibition mainly depends on the surface conditions and mode of adsorption of the inhibitor. Assumptions were made that the uncovered parts of the metal surface is equal to zero and the corrosion process takes place only at the uncovered parts of the metal surface. The degree of surface coverage (θ) has been calculated using θ = IE/100, by assuming a direct relationship between surface coverage and inhibition efficiency, as presented in Table 7. In order to gain an insight to the mode of adsorption of the extract onto the mild steel surface, the surface coverage values were fitted into few adsorption isotherms and the values of correlation coefficient were used to determine the best fit. Adsorption isotherms are usually used to describe the adsorption process. The adsorption isotherm provides important clues regarding the nature of the metal-inhibitor interaction. Inhibitor molecules adsorb on the metal surface if the interaction between molecule and metal surface is higher than that of the water molecule and the metal surface. The most frequently used isotherms include Langmuir, Temkin, Frumkin, Flory-Huggins,.. according to the following equations 8, 9 and 10 [20, 37-45].
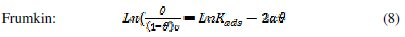
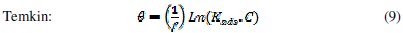
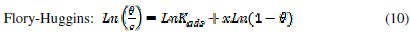
The Frumkin adsorption isotherm (θ vs. C) assumes that there is some interaction between the adsorbate, and the Temkin adsorption isotherm (θ vs. lgC) represents the effect of multiple layer coverage [43] where C is the concentration of the extract, Kads is the adsorptive equilibrium constant, f is the heterogeneous factor of the metal surface describing the molecular interactions in the adsorption layer and the heterogeneity of the metal surface. If f > 0, mutual repulsion of molecules occurs, and if f < 0 attraction takes place. x is the number of inhibitor molecules occupying an active site or the number of water molecules replaced by one molecule of the extract. Kads is related to the standard free energy of adsorption, ΔGads, by the following equation 11, where the numeral of 55.5 is the molar concentration of water in solution, R is the universal gas constant (8.314 J mol-1 K-1) and T is the temperature (K) [46, 48]:

Langmuir adsorption isotherm is given by the following equation 12 [58]:

The rearrangement gives the following equation 13:

The Langmuir isotherm (C/θ vs. C) assumes that there is no interaction between adsorbed molecules on the surface. Langmuir isotherm was tested for its fit to the experimental data. The plots of C/θ versus C which is the Langmuir adsorption isotherm gives a straight line with an intercept of 1/Kads, as shown in Fig. 9 and 10.
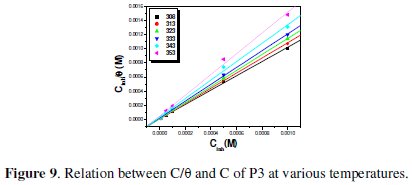
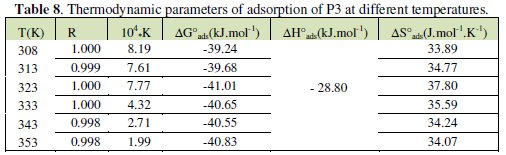
The negative values of DG°ads for P1, P2 and P3 ensure the spontaneity of the adsorption process and stability of the adsorbed layer on the steel surface. Fig. 7 shows the relationship between C/θ and C of P3 at various temperatures. The more negative value of ΔG°ads for P3 indicated that this last one is more strongly adsorbed on the steel surface. Generally, values of DG°ads around -20 kJ mol-1 or lower are consistent with the electrostatic interaction between the charged molecules and the charged metal (physisorption); those around -40 kJ mol-1 or higher involve charge sharing or transfer from organic molecules to the metal surface to form a coordinate type of bond (chemisorption). The calculated DG°ads values of slightly more negative than -40 kJ.mol-1 indicate, therefore, that the adsorption mechanism of the pyridines tested on steel in 1 N HCl solution was typical of chemisorption (Table 8).
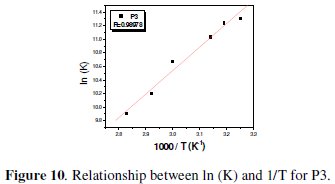
The unshared electron pairs in nitrogen may interact with d-orbitals of steel to provide a protective chemisorbed film [34]. According to the Van't Hoff equation 14 [27]:

To obtain the adsorption heat, the linear regression between ln(K) and 1/T was dealt. Under the experimental conditions, the adsorption heat could be approximately regarded as the standard adsorption heat (ΔHads). ΔH°ads found by the Van't Hoff equation may be also evaluated by the Gibbs-Helmholtz equation, which is defined as follows eq. 15:
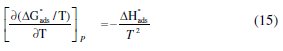
which can be arranged to give the following equation 16:

According to the thermodynamic basic equation 17:

the standard adsorption entropy ΔS°ads could be calculated. All the obtained thermodynamic parameters are shown in Table 8.
The positive values of ΔH* reflect that the process of adsorption of the inhibitors on the steel surface is an endothermic process. In the present case, the standard adsorption heat -28.80 kJ mol-1 shows that a comprehensive adsorption (physical adsorption) might occur. The same results were obtained in previous works [30]. In our study, the negative values of ΔH°ads also show that the adsorption of inhibitor is an exothermic process [20].
On the other hand, values of ΔS°a are more positive in acid solutions containing inhibitors than those obtained in the uninhibited solutions. This behaviour can be explained as a result of the replacement process of water molecules during the adsorption of inhibitors on steel surface. This observation is in agreement with the findings of other workers [5, 6, 14] . However, mild steel corrosion in the free acid was characterized by the more negative ΔS°, a value which implies that the activation complex in the rate determining step required association rather than dissociation [5, 8]. The adsorption of inhibitor molecules was commonly believed that simple adsorption on bare surface was generally an endothermic process which was accompanied by a decrease in entropy.
Adsorption of the organic compound depends upon the charge and the nature of the metal surface, electronic characteristics of the metal surface on adsorption of solvent and other ionic species, temperature of the corrosion reaction and the electrochemical potential at the metal solution interface. The first one is weak undirected interaction due to electrostatic attraction between inhibiting organic ions or dipoles and the electrically charged surface of the metal. This type of interaction involves charge sharing or charge transfer from adsorbate to the atoms of the metal surface in order to form a coordinate type bond and the interaction is termed chemical adsorption or chemisorption [53].
Quantum chemical study
Geometry optimization is a name for the procedure that attempts to find the configuration of minimum energy of the molecule. The procedure calculates the wave function and the energy at a starting geometry and then proceeds to search a new geometry of a lower energy. This is repeated until the lowest energy geometry is found. The procedure calculates the force on each atom by evaluating the gradient (first derivative) of the energy with respect to atomic positions. Sophisticated algorithms are then used at each step to select a new geometry, aiming for rapid convergence to the geometry of the lowest energy. In the final minimum energy geometry, the force on each atom is zero. It is important to recognize that this procedure will not necessarily find the global minimum, i.e., the geometry with the lowest energy. By its nature, a successive search for a minimum finds a local minimum but not necessarily the lowest. In fact, the optimization procedure stops when it finds a stationary point, i.e., a point where forces on the atoms are zero, and this may be also a saddle point (i.e., a transition structure). This will occur particularly if we restrict the symmetry of the molecule and do not allow the program to search the full space of molecular configurationally degrees of freedom. It is always a good idea to start a procedure of a geometry optimization calculation with a small basis set and a relatively poor method before we move to the basis set and method of choice for a particular problem. You can then start the final geometry optimization from the geometry selected by the simpler and less accurate approach. We can even use the second derivatives of the energy with respect to atomic coordinates (obtained during the optimization process or from a frequency run to further improve the geometry optimization) [15].
Complete geometrical optimizations of the investigated molecules are performed using DFT (density functional theory) with the Beck's three parameter exchange functional along with the Lee-Yang-Parr nonlocal correlation functional (B3LYP) [12-14] with 6-31G* basis set is implemented in Gaussian 03 program package [15]. This approach is shown to yield favourable geometries for a wide variety of systems. This basis set gives good geometry optimizations. The geometry structure was optimized under no constraint. The following quantum chemical parameters were calculated from the obtained optimized structure: several quantum parameters will be determined for possible relations with the inhibitor efficiencies of the pyridine molecules (Tables 9 and 10) [16, 17].
The energy of HOMO is often associated with the electron donating ability of a molecule. The less negative HOMO energy and the smaller energy gap (ΔE= ELUMO -EHOMO) are often interpreted by a stronger chemisorption bond and perhaps greater inhibitor efficiency. The calculations were carried out for the pyridine derivatives. The geometry of all compounds under investigation was determined by optimizing all geometrical variables without any symmetry constraints. The harmonic frequencies were computed from analytical derivatives for all species in order to define the minimum-energy structures. According to DFT method, the ionization potential, I can be approximated as the negative of the highest occupied molecular orbital (HOMO) energy [18],

The negative of the lowest unoccupied molecular orbital (LUMO) energy is similarly related to the electron affinity A. In modern terms, we relate affinity to the phenomenon whereby certain atoms or molecules have the tendency to aggregate or bond.

The electron donating power of a donor molecule is measured by its ionization potential which is the energy required to remove an electron from the highest occupied molecular orbital. The overall energy balance (ΔE), i.e., energy gained or lost, in an electron donor-acceptor transfer is determined by the difference between the acceptor's electron affinity (A) and the ionization potential (I):

In chemistry, the class of electron donors that donate not just one, but a set of two paired electrons that form a covalent bond with an electron acceptor molecule, is known as a Lewis base. This phenomenon gives rise to the wide field of Lewis acid-base chemistry [1]. The driving forces for electron donor and acceptor behaviour in chemistry are based on the concepts of electro positivity (for donors) and electronegativity (for acceptors) of atomic or molecular entities. Natural bond orbital (NBO) analysis was performed to evaluate the electron-density distributions. The electron density plays an important role in calculating the chemical reactivity parameters. The global reactivity includes electronegativity, which is identified in the finite difference approximation as the negative of the chemical potential. The ionization energy is typically specified in electron Volts (eV) and refers to the energy required to remove a single electron from a single atom or molecule

In 1983 Pearson together with Robert Parr extended the qualitative HSAB theory with a quantitative definition of the chemical hardness (η) as being proportional to the second derivative of the total energy of a chemical system with respect to changes in the number of electrons at a fixed nuclear environment:

The factor of one-half is arbitrary and often dropped as Pearson has noted. An operational definition for the chemical hardness is obtained by applying a three- point finite difference approximation to the second derivative [19]:
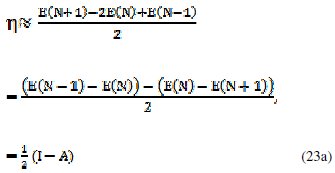
the global hardness (η) defined as

where I is the ionization potential and A the electron affinity. This expression implies that the chemical hardness is proportional to the band gap of a chemical system, when a gap exists.
The first derivative of the energy with respect to the number of electrons is equal to the chemical potential, μ, of the system,

from which an operational definition for the chemical potential is obtained from a finite difference approximation to the first order derivative as
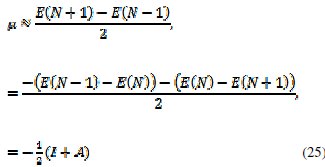
which is equal to the negative of the electronegativity (χ) definition on the Mulliken scale: μ = -χ. The hardness and Mulliken electronegativity are related as:

and in this sense hardness is a measure for resistance to deformation or change. Likewise a value of zero denotes maximum softness, where softness is defined as the reciprocal of hardness and the global softness (σ) as its inverse:

During the interaction of the pyridine derivatives with the steel surface, electrons flow from the lower electronegativity pyridine derivative to the higher electronegativity steel surface until the chemical potential becomes equalized. The fraction of the transferred electron, DN was estimated according to Pearson [15, 18, 20]:

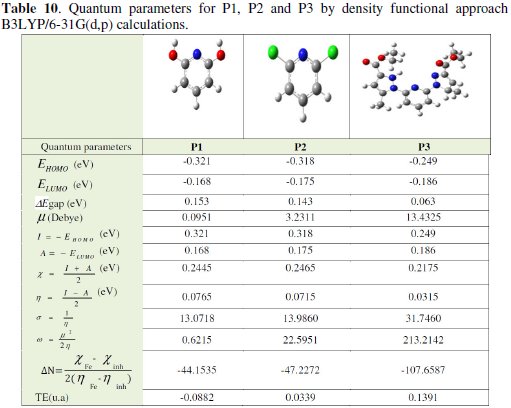
Quantum chemical parameters obtained from the calculations which are responsible for the inhibition efficiency of inhibitors, such as the highest occupied molecular orbital (EHOMO), energy of lowest unoccupied molecular orbital (ELUMO), HOMO-LUMO energy gap (ΔEH-L), dipole moment (μ) and total energy (TE), electronegativity (χ), electron affinity (A), global hardness (η), softness (σ), ionization potential (I), the global electrophilicity (ω), the fraction of electrons transferred from the inhibitor to steel surface (ΔN) and the total energy (TE), are collected in Table 10.
In Fig. 11, we have presented the frontier molecule orbital density distributions of the studied compounds.
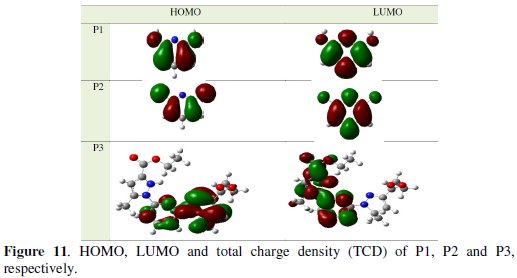
Analysis of Fig. 11 shows the distribution of two energies HOMO and LUMO; we can see that the electron density of the HOMO location in the pyridine molecules is mostly distributed near the nitrogen (N) atoms and pyridine rings, indicating that these are the favourite sites for adsorption, while the density LUMO was distributed almost of the entire molecules.
The EHOMO is a quantum chemical parameter which is often associated with the electron donating ability of the molecule. High value of EHOMO = -0.249 eV for P3 is likely to a tendency of the molecule to donate electrons to appropriate acceptor molecule of low empty molecular orbital energy and indicates also the better inhibition efficiency [15, 17].
It has also been found that an inhibitor does not only donate an electron to the unoccupied d orbital of the metal ion but can also accept electrons from the d orbital of the metal leading to the formation of a feedback bond. Therefore, the tendency for the formation of a feedback bond would depend on the value of ELUMO. The lower the ELUMO, the easier is the acceptance of electrons from the d orbital of the metal. Based on the values of ELUMO, the order obtained for the decrease in inhibition efficiency (P3 < P2 < P1) was also similar to the one obtained from experimental results.
A band gap, also called an energy gap, is an energy range in a solid where no electron states can exist. In graphs of the electronic band structure, the band gap generally refers to the energy difference (in electron Volts) between the top of the valence band and the bottom of the conduction band in metal. This is equivalent to the energy required to free an outer shell electron from its orbit about the nucleus to become a mobile charge carrier, able to move freely within the solid material, so the band gap is a major factor determining the electrical conductivity of a metal.
We concluded that the large values of the energy gap (ΔE) imply high electronic stability and then low reactivity, when low values imply that it will be easier to remove an electron from the HOMO orbital to LUMO one which can result in good inhibition efficiency. As ΔE decreases, the reactivity of the molecule increases leading to increase the inhibition efficiency of the molecule.
The results presented in Table 10 show that ΔEgap = ELUMO -EHOMO = 0.063 eV for P3 have the lowest energy gaps. Therefore, their higher reactivity can allow them to be easily adsorbed onto the mild steel surface leading to increase their inhibitive efficiencies compared to P2 ΔEgap = 0.143 eV [21].
Dipole moment is the measure of polarity of a polar covalent bond. It is defined as the product of charge on the atoms and the distance between the two bonded atoms. The dipole moment of the molecule gives information on the polarity of the given system, however, reflects only the global polarity of a molecule. For a complete molecule the total molecular dipole moment may be approximated as the vector sum of individual bond dipole moments.
The dipole moment (μ in Debye) is another important electronic parameter that results from non-uniform distribution of charges on the various atoms in the molecule. Molecules that have high dipole moment have a tendency to interact with other molecule through electrostatic interactions (e.g., dipole-dipole interactions). The high value of dipole moment probably increases the adsorption between the chemical compound and the metal surface [15]. The energy of the deformability increases with the increase in μ, making the molecule easier to adsorb at the steel surface. The volume of the inhibitor molecules also increases with the increase of μ. This increases the contact area between the molecule and surface of steel, increasing the corrosion inhibition ability of the inhibitors.
The dipole moment of a molecule is strongly determined by the shape of the molecule, size of the molecule and type of atoms constituting the molecule. The relationship between inhibition efficiencies and dipole moment of similar molecules have often given results that are not univocal, i.e., in some instances the dipole moment appears to increase with increasing inhibition efficiencies, while in other systems the dipole moment appears to decrease with increase in the inhibition efficiencies of the inhibitors. In our study, there is direct relationship between the EI(%) and the dipole moment. The results of the dipole moment for P2 and P3 are reported in Table 10. The dipole moment follows the order; μ= 13.4325 Debye for P3 > μ= 3.2311 Debye for P2, which is not in agreement with trends in the experimental inhibition efficiencies of the inhibitors earlier reported in literature.
The total energy presented in Table 10, calculated by quantum chemical methods is also a beneficial parameter. The total energy of a system is composed of the internal, potential, and kinetic energy. The total energy of a system including that of the many body effects of electrons (exchange and correlation) in the presence of static external potential (for example, the atomic nuclei) is a unique functional of the charge density. The minimum value of the total energy functional is the ground state energy of the system. The electronic charge density which yields this minimum is then the exact single particle ground state energy. In our study the total energy of the best inhibitor P3 is equal to 0.13917 a.u.; the value of compound P2 is equal to 0.0339 a.u.
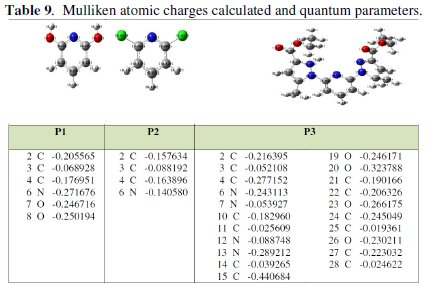
Mulliken charges showed in Table 9, arise from the Mulliken population analysis and provide a means of estimating partial atomic charges from calculations carried out by the methods of computational chemistry, particularly those based on the linear combination of atomic orbital molecular method. The Mulliken charges obtained using DFT/B3LYP/6-31G are significantly different from the results of other methods. This result is consistent with the fact that Mulliken atomic charges are strongly basis-set sensitive and in most instances tend to become unphysical when large basis sets with diffuse functions are employed [22]. The Mulliken atomic charges obtained are also not realistic. The partial charges on the individual atoms in a molecule also indicate the reactive centres for a particular inhibitor. Atoms with the highest negative charge are considered to have an electron donor role when interacting with metal surfaces. The Mulliken atomic charges for the heteroatoms of the pyridine derivatives are reported in Table 9 and show that N atoms have the highest negative charge. The partial charges on the individual atoms in a molecule also indicate the reactive centres for a particular inhibitor. Atoms with the highest negative charge are considered to have an electron donor role when interacting with metal surfaces. Moreover, the negative charge on the N atom increases across the structures, respectively for P3 and (P1, P2).
This result is consistent with the fact that the electron withdrawing effect of some N atoms in the structure for P3, and decreases the negative charge on the N atom in the ring while the electron donating role. In view of the above results, P3 has greater tendency to adsorb on the metal surface because it has three highly negative charged centres, while P2 is preferentially the molecule with the least negative charge.
Absolute hardness and softness are important properties to measure the molecular stability and reactivity. It is apparent that the chemical hardness fundamentally signifies the resistance towards the deformation or polarization of the electron cloud of the atoms, ions or molecules under small perturbation of chemical reaction. A hard molecule has a large energy gap and a soft molecule has a small energy gap [23]. Soft molecules are more reactive than hardness because they can easily offer electrons. Hence, inhibitors with the highest values of global softness (the least values of the global hardness) are expected to be good corrosion inhibitors for bulk metals in acidic media. For the simplest transfer of electron, adsorption could occur at the part of the molecule where softness (σ), which is a local property, has a highest value. In our present study P3 with low softness value σ = 31.7460(eV) compared with other compounds has a low energy gap. The most widely used quantity to describe the polarity is the dipole moment of the molecule.
The fraction of electrons transferred describes the trend of electrons donation within a set of inhibitors. In literature it has been reported that the values of ΔN show inhibition effect resulted from electrons donation. According to our study, the inhibition efficiency increased with increasing the electron donating ability of the inhibitor at the metal surface and increasing electron-donating ability at the metal surface.
It is worth noting that all the computed descriptors are in total agreement with the experimental inhibition efficiencies of the investigated inhibitors, which confirms that the presence of heteroatoms enhances the donor abilities of these molecules increasing their adsorption on the metal surface and thereby leading to higher experimental inhibition efficiencies.
Conclusion
The results of the present work can lead to the following conclusions:
1. The organics tested reveal excellent protective qualities for protection of mild steel against corrosion in 1N HCl medium. This results in an increase in inhibition efficiency. This increase is particularly marked when the concentration is high and attains around 92% for P3.
2. Reasonable good agreement was observed between the data obtained from potentiodynamic polarization and electrochemical impedance spectroscopy techniques.
3. P1, P2 and P3 slows down the cathodic reaction to greater extent than the anodic one, suggesting that these compounds mainly act as mixed-type inhibitors.
4. The adsorption of all inhibitors is well described by the Langmuir adsorption isotherm. All values of the standard Gibbs energy of adsorption are negative, confirming that both chemisorption and physisorption control the adsorption phenomenon.
5. The study of the influence of temperature in the range between 298 and 333 K showed that the inhibition rate decreases with temperature and the activation energy is modified by the presence of inhibitors. The change could be attributed to the chemisorptions of the inhibitor to the surface of the steel.
6. The impact of concentration, temperature and immersion time taken together on corrosion inhibition process demonstrates the double character nature of the adsorption phenomena. It remains not evident to attribute bluntly either chemisorption or physisorption mechanism for the studied inhibitors. The authors believe that the studied inhibitors are adsorbed by mixed mode (physisorption and chemisorption both involved to some extent) onto the mild steel interface.
7. The electrochemical impedance results show that the Nyquist diagrams obtained have a single capacitive loop indicating that the corrosion of mild steel is mainly controlled by the charge transfer process.
8. This study has also suggested that the inhibitors act by adsorption by forming a protective layer on the metal surface and the piperidine tested adsorbs according to the Langmuir isotherm. Determination of heat of adsorption reinforced the chemisorption postulate.
9. Theoretical calculations (quantum chemical) are in good agreement with the results obtained from electrochemical study and structure-corrosion protection relationships, which also confirms that the adsorption centre is N atoms.
References
1. Barouni K, Bazzi L, Albourine A. Annales De Chimie-Science Des Materiaux. 2010;35:333. [ Links ]
2. El Ouali I, Chetouani A, Hammouti B, Aouniti A, Touzani R, El Kadiria S and Nlate S. Port Electrochim Acta. 2013;31:53.
3. Abiola OK, John MO, Asekunowo PO, Okafor PC, James OO. Green ChemLett Rev. 2011;4:273.
4. Beenakumari KS. Port Electrochim Acta. 2013;31:235-239. [ Links ]
5. Chetouani A, Daoudi M, Hammouti B, et al. Corros Sci. 2006;48:2987. [ Links ]
6. Bouklah M, Attayibat A, Kertit S, et al. Appl Surf Sci. 2005;242:399. [ Links ]
7. Musa AY, Kadhum AAH, Mohamad AB, et al. J Central South Univ Technol. 2010;17:34. [ Links ]
8. Bentiss F, Jama C, Mernari B, et al. Corros Sci. 2009;51:1628. [ Links ]
9. Zarrok H, Zarrouk A, Salghi S, et al. Port Electrochim Acta. 2012;30:405. [ Links ]
10. Figueira RM, Pereira EV, Silva CJR, et al. Port Electrochim Acta. 2013;31:277. [ Links ]
11. Musa AY, Kadhum AAH, Takriff MS, et al. Corros Eng Sci Technol. 2010;45:163. [ Links ]
12. Keles H, Keles M, Dehri I, et al. Mater Chem Phys. 2008;112:173. [ Links ]
13. Chetouani A, Hammouti B, Aouniti A, et al. Prog Org Coat. 2002;45:373. [ Links ]
14. Ouchrif A, Zegmout M, Hammouti B, et al. Prog Org Coat. 2005;53:292. [ Links ]
15. Zarrouk A, Zarrok H, Salghi R, et al. Int J Electrochem Sci. 2012;7:6353. [ Links ]
16. Bouklah M, Harek H, Touzani R, et al. Arab J Chem. 2012;5:163. [ Links ]
17. Boussalah N, Ghalem S, El Kadiri S, et al. Res Chem Intermed. 2012;38:2009. [ Links ]
18. Ebenso EE, Kabanda MM, Arslan T, et al. Int J Electrochem Sci. 2012;7:5643. [ Links ]
19. Eddy NO, Ameh PO, Gwarzo MY, et al. Port Electrochim Acta. 2013;31:79. [ Links ]
20. Zarrouk, Hammouti B, Zarrok H, et al. Int J Electrochem Sci. 2012;7:89. [ Links ]
21. El Ashry EH, El Nemr A, Ragab S. J Molec Model. 2012;18:1173. [ Links ]
22. Zarrok H, Zarrouk A, Salghi R, et al. Int J Electrochem Sci. 2012;7:8958. [ Links ]
23. Zarrok H, Al-Deyab SS, Zarrouk A, et al. Int J Electrochem Sci. 2012;7:4047. [ Links ]
*Corresponding author. E-mail address: hammoutib@gmail.com
Received 4 March 2014; accepted 4 April 2014

