










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.32 no.1 Coimbra jan. 2014
https://doi.org/10.4152/pea.201401021
Tuning the Initial Electronucleation Mechanism of Palladium on Glassy Carbon Electrode
Tibebu Alemu , Birhanu D. Assresahegn and Tesfaye R. Soreta*
Jimma University, College of Natural Sciences, Department of Chemistry, P.O. Box 378, Jimma, Ethiopia
Abstract
Electrochemical metal nucleation is the method for the formation of metal nanoparticles on the electrode surface. Studying the early stage of electronucleation is simpler than other methods as the driving force for nucleation is achieved by changing deposition potentials and concentration of metal ions. In this work, the potential step electrochemical deposition of palladium was studied from its chloride solution at room temperature on glassy carbon electrode surface. The nucleation mechanism was studied by analysis of the resulting current transients. Accordingly, the initial electro-nucleation mechanism of palladium nanoparticles was found to be varying depending on deposition conditions such as deposition potential and palladium concentration. It can be changed from 3D instantaneous (for all deposition potentials studied and in higher electrolytic concentration) to 3D progressive nucleation mechanism (for lower deposition potential and lower electrolytic concentration). In addition, the nucleation rate for each deposition potential as well as the concentration has been determined. The nucleation rate in this research is used to calculate the nuclei density and found to decrease from more negative deposition potential to more positive deposition potential in agreement with the observed shift in electronucleation mechanism.
Keywords: nanoparticles, palladium, initial nucleation, electrodeposition, glassy carbon electrode.
Introduction
Metal nanoparticles are expected to play a major role in future nanotechnologies. The physical and chemical properties of the nanosized materials differ significantly from their bulk counterparts as a result of small size effects. The properties of deposited metal nanoparticles on substrates are in turn strongly determined by the final surface structure which is also determined by the early stages of nucleation [1, 2]. The surface structure and properties of deposited metal nanoparticles are governed by a competition between the rate of nucleation and the crystal growth [1]. For the conditions that favor the rapid formation of crystal nuclei at initial stage of deposition, the surface structure of deposited metal will be fine grained and highly applicable in nanotechnology. However, if the conditions favor the nuclei to grow rapidly, relatively large crystals will be formed and the deposited structure becomes rough in appearance and less applicable in nanotechnology. Thus, the detailed knowledge of such correlations is helpful for optimizing metal nanoparticles fabrication processes. Electrochemical nucleation of metals is an easy, rapid, flexible and effective tool for structuring and modification of solid state surfaces [1, 3-5]. Electrochemical nucleation is a phase formation process in which an electric current passing through an electrochemical cell from an external source causes a redox reaction (especially reduction of electro active species) resulting in the formation of deposited atoms on an electrode surface. This technique allows the formation of several grain sizes on which many chemical and physical properties of nanostructured materials depend. The deposit exhibits surface structure without domination by voids which is a major limitation to other deposition methods such as physical vapor deposition methods. Due to these advantages, electrodeposition becomes greatly significant for the development of micro and nano system technologies. On an electrode surface, the electrochemical deposition process involves at least three steps: (a) transfer of hydrated metal ions or complexes from bulk of solution to the electrode-electrolyte interphase, (b) adsorption of this metal ions/complex on the surface, and finally (c) charge transfer at the surface leads to the reduced atom [2]. Metal deposition and dissolution takes place at defects which act as the preferred sites for nucleation and growth on a substrate since these are active sites (step and kink sites). The final size distribution and property of the electrodeposits, strongly depends on the nucleation and growth mechanism. The nucleation mechanism is again influenced by the presence of organic additives, applied overpotential and the nature of the substrate. Depending on the above factors, an initial stage of electronucleation involves either an instantaneous or a progressive nucleation mechanism [6-10]. In the case of instantaneous nucleation mechanism, all the electrode active sites are occupied by the nuclei at early stages. The nuclei formed are widely spaced and the crystals grow as time of deposition prolonged with non overlapping (individual nuclei size increased without increased in nuclei density) as there is no interaction between each nucleus. However, the number of nuclei that are formed is a function of electrodeposition time in the progressive nucleation mechanism. In such cases, early established nuclei gradually grow and overlap while other new nuclei are formed. Therefore, the progressive nucleation process exhibits zones of reduced nucleation rate around the growing stable nuclei [11, 12].
The study of initial electrocrystalization of metals is an important area of study [7-10, 13-35]. A number of studies have been reported for the investigation of the initial nucleation of cobalt [32], copper [14, 22, 25, 27, 29, 33], palladium [7, 13, 16-19, 21, 23, 34, 36], platinum [20, 28] and silver [31] on various substrates. Initial electro-nucleation studies are important as it provides a mechanistic study with simple approach that can be used to approximate the actual initial nucleation without the help of expensive instruments such as atomic force microscopy and other nano-imaging tools.
Palladium is a well known catalyst for many reactions such as electrocatalytic oxidation of formaldehyde [37, 38], hydrogenation of unsaturated hydrocarbons [39], oxidation of alcohols, and is routinely used in the automobile industry in catalytic converters to reduce the amounts of nitrogen oxides, carbon monoxide, and un-burned hydrocarbons [40].
Palladium is used in electronic industry, in view of its excellent resistance to corrosion and wear, good solderability and lower density, in the development of biosensors, and has been exploited as a component in an enzymatic glucose biosensor [41] as well as for detection of catecholamine [42] and DNA [43]. The initial electronucleation of a metal is dependent on parameters such as type and source of metal ion, concentration of metal ion, electrolyte and the substrate on which deposition takes place. Hence, with the variation of these parameters, independet independent study has to be carried out to understand the mechanism the initial nucleation. In this paper, a comprehensive investigation was performed to understand the initial electrochemical nucleation process of palladium (Pd) on glassy carbon electrode (GCE). The effects of overpotentials and concentrations of palladium precursor on the initial nucleation and growth mechanisms of Pd were investigated. In addition, nucleation rate at the studied conditions was calculated to gain deeper insights of the electrodeposition process.
Experimental
Chemicals
Palladium dichloride (PdCl2, 99.9%, Aldrich), potassium nitrate (KNO3, 99%, Nice), potassium hexacyanoferrate (K3[Fe(CN)6], 97%, Labmerk Chemicals) and sodium perchlorate (NaClO4, 98%, Aldrich), citric acid (C6H8O7, 99.5%, Wardle Chemicals LTD), hydrochloric acid (HCl, 37%, Riedel-de Haen), sodium chloride (NaCl, 99.8%, Uni-Chem), potassium hydroxide (KOH, 96%, BDH) were used for this work as recieved. Double distilled water was used to prepare all aqueous solutions.
Instrumentation
Electrochemical measurements
Cyclic voltammetry and amperometric experiments were carried out using BAS Epsilion EC-Version 1.40.67 voltammetric analyzer (Bio-analytical Systems, USA) controlled with basic epsilon software. A conventional three-electrode setup was used with a 3 mm diameter glassy carbon electrode (GCE, BASi, MF 2012) as the working electrode and a platinum wire counter electrode (BASi, MW-1032). An Ag/AgCl electrode (BASi MF-2042) served as a reference electrode. All potentials were reported with respect to this reference electrode. The solution of PdCl2 was stirred with a magnetic stirrer during palladium deposition using BASi C3 Cell Stand at 500 rpm. All experiments were conducted at room temperature.
Methods
Solution preparation
For the study of Pd concentration effects on nucleation mechanism, a 7.5 mM PdCl2 deposition bath was prepared by adding 6.65 mg of PdCl2 to a 50 mL of pH 2 citrate buffer. The solution was homogenized by shaking and left overnight in the dark after which a clear wine-red solution was obtained [44]. Finally, the pH of the solution was adjusted to 3 by addition of diluted KOH. This solution was used as stock solution to prepare other lower concentrations of PdCl2. For electrode area determination, a 0.1 mM K3[Fe(CN)6] in 0.1 M NaClO4 was used.
Electrode preparation
The cleaning procedure for the GCE included polishing and electrochemical conditioning steps similar to the procedure reported by Soreta et al. [44]. For polishing, 0.05 μm alumina slurry (BASi CF-1050) was used. Upon polishing, the electrodes were carefully rinsed with deionized water. Then, the electrodes were conditioned by potential scanning from 0 V to 1.4 V in 1 M NaClO4 for at least six complete scans at 50 mV/s, where the high background current due to glassy carbon oxidation diminished, and a reproducible cyclic voltammogram was obtained. The background current of the bare electrode was measured by cyclic voltammetry within the potential window of 0 to 0.7 V. Electrodes with a high background current above a selected reference were re-polished and conditioned. The electrodes were used immediately following the cleaning and conditioning steps.
Determination of electrode area
The real electrode area of the bare GCE was determined by chronocoulometry for duration of 250 milliseconds. It was estimated in 0.1 M NaClO4 using 0.1 mM potassium hexacyanoferrate (K3[Fe(CN)6], diffusion coefficient 7.6 × 10-6 cm2 s-1) using Anson's equation [45].
Electrodeposition of palladium and study of nucleation mechanism
Pd was electrodeposited on GCE from the deposition bath, by applying the potentials from the rest potential of 1.1 V to all deposition potentials studied for duration of 1 s to determine nucleation models. The palladium deposition bath concentrations studied were 0.75 M, 1.5 M and 7.5 M. The nucleation mechanisms were studied using mathematical models developed by Scharifker and Hills [11, 12]. The experimental current transients obtained at different deposition potentials were inserted to those equations, and dimensionless current (i/imax)2 with their corresponding instantaneous as well as progressive values were calculated. Then the values obtained from experimental dimensionless current versus dimensionless time (t/tmax) were plotted using Microcal origin software. Finally they were compared to each other for determination of nucleation mechanism whether the experimental curves best fit to instantaneous or progressive. The nucleation rate also was calculated using their corresponding mathematical equations which have been previously developed [46] to explain the nucleation rate.
Results and discussion
Cyclic voltammetry
The cyclic voltammogram for the electrochemical deposition of Pd from the deposition bath is given in Fig. 1.
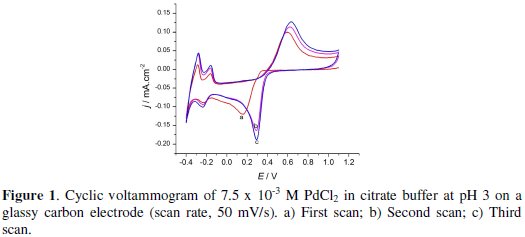
The potential scan was started at +1.1 V. Palladium started to deposit at +0.3 V for the first reduction scan and at +0.6 V for the second and third scans.
The cathodic peak at +0.15 V for the first scan is due to the palladium deposition. The peak observed at -0.20 V in all scans is due to hydrogen adsorption to the electrode surface resulting in PdHx [44]. At the potentials more negative than 0.30 V, the peak observed is due to hydrogen evolution. On the return scan, hydrogen is stripped off at a potential of -0.30 V, while the peak at -0.15 V observed next to hydrogen oxidation was due to the desorption of already adsorbed hydrogen, and finally palladium started dissolution from the electrode at +0.38 V. The residual anodic current observed at the anodic limit suggests the passivation of the Pd particles [47]. As a result for the second and third voltammetric cycle a lower overpotential is required to initiate the nucleation and growth of Pd deposits. The cross over indicates that there is a formation of nucleation on GCE. The general behavior of the CV depicted in Fig. 1 is in good agreement with the one reported by Soreta et al. [44].
Nucleation and growth of palladium
Current-time transients
In order to analyze the nucleation and growth processes of electrochemical deposition, either of the two mechanisms, charge-transfer-controlled or masstransfer- controlled, has to be suppressed. A set of potentiostatic current transients was obtained by stepping the potential, with chronoamperometric technique, from E = 1.1 V to different potential values E, ranged between 0.60 ≤ E /V ≤ 0.15, that can make the deposition process mass-transfer-controlled according to the voltammogram recorded, for 1 s in stirred citrate pH 3 buffered solution (Fig. 2).
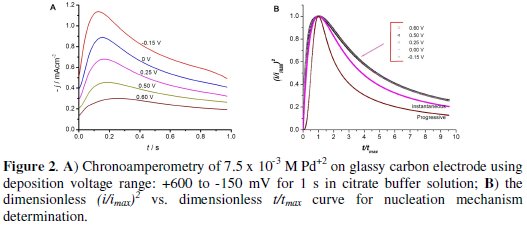
Initially, the current increases due to the formation and growth of palladium nanoparticles. At later stages of the nucleation process the individual diffusion zones grow and the current reaches a maximum peak and then decays. This behavior indicates that the reaction kinetics is controlled by diffusion of electroactive ions to the electrode surface [47].
The trend in these transients indicates that the Pd deposition follows the three- dimensional nucleation mechanism with mass-transfer-controlled growth [47]. The maximum current (Imax) and the time at which Imax appears (tmax) change as a function of the applied potential.
Analysis of current-time transients
The current-time transients was analyzed using the mathematical model that Scharifker and Hills have derived (here after, SH theory) [12]. The SH theory describes the three-dimensional nucleation process based on the current-time transients. According to the SH theory, the mathematical expressions in a non- dimensional form are given by:
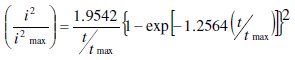
for instantaneous nucleation, and
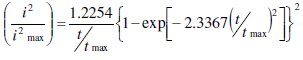
for progressive nucleation [11, 12].
Fig. 2 depicts the (I/Imax)2 versus (t/tmax) plots of the experimental data at the different potentials for the analysis of the nucleation and growth mode of Pd on GCE.
The initial nucleation mechanism from all experimental plots representing the dimensionless current transient best describes the three-dimensional hemispherical instantaneous nucleation mechanism. Further increase in the deposition time would only increase the size of the nanocrystals and not their number density because no new nucleation sites are created.
Although some experimental data are deviated from the theoretical lines at longer times (t/tmax > 1), these deviations are often reported in the literature [48]. Radisic et al. [48] demonstrated that the hydrogen evolution could induce such deviations. We believe that the adsorption of hydrogen ions can also cause deviations during the Pd deposition process with large overpotentials. However, the reasons for the deviations from the SH theory have not yet been elucidated thoroughly.
Effect of concentration on the nucleation mechanism of palladium
The concentration effects were studied using 0.75 M, 1.50 M, and 7.5 × 10-3 M Pd+2 solutions in citrate buffer solution (pH 3) at two different and previously studied deposition potentials,-150 mV and 600 mV. The results are presented in Fig. 3.
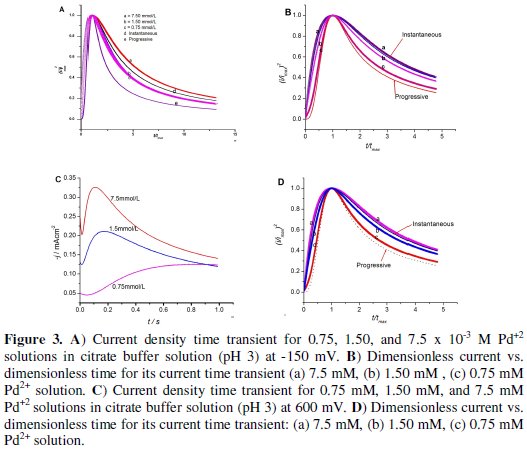
According to the result observed from the graphs below, the nucleation mechanism was instantaneous for -150 mV up to t/tmax being equal to 1, but as the reaction proceeds, it slightly deviated from instantaneous with increasing concentration of palladium ion which can be attributed to the adsorption of hydrogen.
The nucleation models observed for 600 mV, Fig. 3 (b), were different from the first one depicted in Fig. 2 (a). In fact it was progressive at low concentration but changed to instantaneous as the concentration of palladium ion was increased to 1.5 and 7.5 × 10-3 M. This is the most interesting result obtained as it gives an option to tailor the nucleation mechanism of Pd on the GC electrode surface by optimizing the concentration of the metal and the deposition potential as needed. The 600 mV potential could give low current density for 0.75 × 10-3 M Pd2+, which results in slow discharge of ions. The nuclei grow faster before the formation of other new nuclei because this potential is less energetic to produce new nucleation sites as compared to high deposition potential [12]. In general, in these conditions, the rate of growth of the nuclei exceed the rate at which a new one is formed; the deposits obtained under these conditions should be coarsely crystalline. As the current density is raised (1.5 and 7.5 × 10-3 M Pd2+), the rate of formation of nuclei will be greater than the rate of growth of the nuclei (instantaneous nucleation) and hence deposited morphology should appear fine grained [49].
The nucleation rate
The nucleation rate (J) of the metal on the GCE surface was calculated using the equation developed by Scharifker-Hills [11, 12]. This model developes the following equation to calculate the nuclei density, No.
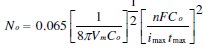
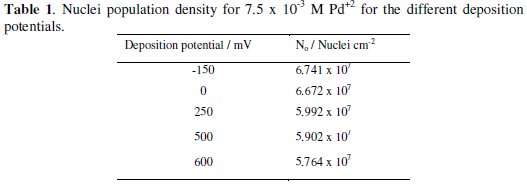
Here, Co is the concentration of metal ion in bulk of solution, Vm the molar volume, F the Faraday constant and n is the number of electrons transferred. And for the same experimental set up in this work, the number density of active nucleation sites for different deposition potentials were calculated and summarized in Table 1.
The time dependence of the number density of nuclei N(t) formed on a substrate exhibiting equally active nucleation sites No has been described as [12, 26]
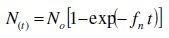
where fn represents the frequency of nucleation per nucleation site. For very high nucleation frequencies, fn, all nucleation sites, No, instantaneously convert to nuclei; each nuclei growth independently, with non-overlapping, and with constant number density throughout the reaction, so that the nucleation rate J = N(t) = No [46], and the process would be instantaneous nucleation. At relatively low fn however, the nucleation sites convert to a nuclei progressively with a stationary nucleation rate J = No fn and the process should be progressive nucleation. In such processes an already formed nuclei will grow by over-lapping with the others and new nuclei will be formed so that there is an increasing in number of nuclei as a function of time but the nucleation rate is small as compared to instantaneous nucleation mechanism [46].
For a three-dimentional hemispherical progressive nucleation mechanism, the nucleation rate can be calculated as [12]:
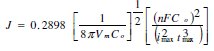
The saturated number density of nuclei Ns corresponding to this nucleation rate and diffusion coefficient can be estimated from the equation below:
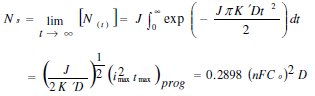
whereas K' is a constant and is equals to 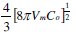 and D is the diffusion coefficient.
and D is the diffusion coefficient.
Based on this SH theoretical models, the effects of different concentrations of Pd on the nuclei population density were evaluated and summarized in Table 2.

In both deposition potentials, when the mechanism of nucleation is instantaneous, the nuclei number density decreases with increasing concentration of the metal. In the case of the progressive nucleation mechanism obtained at a concentration of 0.75 × 10-3 M Pd2+, the nucleation rate J can be extracted from the coordinates of the current maximum using the SH mode [12]. From the transient experimental current maximum, obtained above 600 mV, results a value of J = 3.704 × 108 s-1 cm-2 and a diffusion coefficient equal to 7.412 × 10-6 s-1 cm2. From these values the saturated nuclei number density can be predicted to be 2.47 × 107 nuclei/cm-2. This value is less than No for larger palladium concentration because progressive nucleation has reduced the nucleation rate as compared to instantaneous nucleation.
Conclusions
In this work the initial nucleation mechanism of palladium on GCE was studied. It has been confirmed that the experimental curves fit better to the threedimentional instantaneous nucleation mechanism for all deposition potentials using 7.5 × 10-3 M Pd2+, but it was changed to three-dimentional progressive nucleation mechanism for low concentration of the metal and more positive deposition potentials. This provides an option to taylor the mechanism of initial nucleation and grain size of the nuclei by optimizing only the concentration of palladium and its deposition potentials on the surface of glassy carbon electrode.
References
1. Penner R M. Metal Deposition, Elsevier, 2007. [ Links ]
2. Paunovic M, Schlesinger M. Fundamentals of Electrochemical Deposition. USA: Wiley; 2006. [ Links ]
3. Jovic V D, Jovic B M, Pavlovic M G. Electrochim Acta. 2006;51:5468. [ Links ]
4. Gorer S, Liu H T, Stiger R M, et al. Metal Nanoparticles: Synthesis, Characterization, and Applications. 2002, 237-259. [ Links ]
5. Zoval J V, Lee J, Gorer S, et al. J Phys Chem B. 1998;102:1166. [ Links ]
6. Radisic A, Vereecken P M, Hannon J B, et al. Nano Letters. 2006;6:238. [ Links ]
7. Quayum M E, Ye S, Uosaki K. J Electroanal Chem. 2002;520:126. [ Links ]
8. Milchev A, Staikov G. Indian J Chem. Inorg Bio-Inorg Phys Theor Anal Chem. 2005;44:899. [ Links ]
9. Milchev A, Heerman L. Electrochim Acta. 2003;48:2903. [ Links ]
10. Budevski E, Staikov G, Lorenz W J. Electrochim Acta. 2000;45:2559 [ Links ]
11. Scharifker B R, Mostany J, Palomar-Pardave M, et al. J Electrochem Soc. 1999;146:1005. [ Links ]
12. Scharifker B, Hills G. Electrochim Acta. 1983;28:879. [ Links ]
13. Gimeno Y, Creus A H, Carro P, et al. J Phys Chem B. 2002;106:4232. [ Links ]
14. Schneeweiss M A, Kolb D M. Phys Status Solidi. 1999;173:51. [ Links ]
15. Kibler L A, Kleinert M, Lazarescu V, et al. Surf Sci. 2002;498:175. [ Links ]
16. Hoyer R, Kibler L A, Kolb D M. Electrochim Acta. 2003;49:63. [ Links ]
17. Robach Y, Abel M, Porte L. Surf Sci. 2003;526:248. [ Links ]
18. Kibler L A, Kleinert M, R. Randler R, et al. Surf Sci. 1999;443:19. [ Links ]
19. Kibler L A, Kleinert M, Kolb D M. Surf Sci. 2000;461:155. [ Links ]
20. Andreazza P, Andreazza-Vignolle C, Rozenbaum J P, et al. Surf Coat Tech. 2002;151:122. [ Links ]
21. Tong X Q, Aindow M, Farr J P G. J Electroanal Chem. 1995;395:117. [ Links ]
22. Huang L, Lee E-S, Kim K-B. Colloids Surf A: Physicochem Eng Asp. 2005;262:125. [ Links ]
23. Lau P P, Wong C C, Chan L, et al. J Electrochem Soc. 2004;151:C436. [ Links ]
24. Tian M, Wang J, Kurtz J, et al. Nano Letters. 2003;3:919. [ Links ]
25. Huang J F, Luo H M, Dai S. J Electrochem Soc. 2006;153:J9. [ Links ]
26. Eliaz N, Eliyahu M. Electrochemical processes of nucleation and growth of hydroxyapatite on titanium supported by real-time electrochemical atomic force microscopy. 2007. [ Links ]
27. Lee J J, Miller B, Shi X, et al. J Electrochem Soc. 2001;148:C183. [ Links ]
28. Teranishi T, Hosoe M, Miyake M. Adv Mater. 1997;9:65. [ Links ]
29. Vereecken P M, Strubbe K, Gomes W P. J Electroanal Chem. 1997;433:19. [ Links ]
30. Kibler L A, Kleinert M, Randler R, et al. Surf Sci. 1999;443:19. [ Links ]
31. Li W, Virtanen J A, Penner R M. Appl Phys Lett. 1992;60:1181. [ Links ]
32. Mendoza-Huizar L H, Robles J, Palomar-Pardave M. J Electroanal Chem. 2003;545:39. [ Links ]
33. Milchev A, Zapryanova T. Electrochim Acta. 2006;51:4916. [ Links ]
34. Diculescu V C, Chiorcea-Paquim A M, Corduneanu O, et al. J Solid State Electrochem. 2007;11:887. [ Links ]
35. Penner R M. Metal Deposition. Elsevier;2007. [ Links ]
36. Corduneanu O, Diculescu V C, Chiorcea-Paquim A-M, et al. J Electroanal Chem. 2008;624:97. [ Links ]
37. Gao G-Y, Guo D-J, Li H-L. J Power Sources. 2006;162:1094. [ Links ]
38. Safavi A, Maleki N, Farjami F, et al. J Electroanal Chem. 2009;626:75. [ Links ]
39. Mukherjee D. J Nanoparticle Res. 2008;10:429. [ Links ]
40. Nishihata Y, Mizuki J, Akao T, et al. Nature. 2002;418:164. [ Links ]
41. Santhosh P, Manesh K M, Uthayakumar S, et al. Bioelectrochem. 2009;75:61. [ Links ]
42. Thiagarajan S, Yang R-F, Chen S-M. Bioelectrochem. 2009;75:163. [ Links ]
43. Chang Z, Fan H, Zhao K, et al. Electroanalysis. 2008;20:131. [ Links ]
44. Soreta T R, Strutwolf J, O'Sullivan CK. Langmuir. 2007;23:10823. [ Links ]
45. Anson F C. Anal Chem. 1966;38:54. [ Links ]
46. Abyaneh M Y. J Electroanal Chem. 2002;530:82. [ Links ]
47. Alvarez A E, Salinas D R. Electrochemically Deposited Palladium Nanocrystals on Vitreous Carbon. In 2nd Mercosur Congress on Chemical Engineering4th Mercosur Congress on Process Systems Engineering. Rio de Janeiro, Brazil; 2005. [ Links ]
48. Staikov G, Milchev A. In The Impact of Electrocrystallization on Nanotechnology. Wiley-VCH Verlag GmbH & Co. KGaA;2007. P. 1-29.
49. E. Guaus E, Torrent-Burgues J. Port Electrochim Acta. 2007;25:139.
Acknowledgements
The authors gratefully acknowledge Jimma University, School of Graduate Studies for funding the research project as well as Department of Chemistry of Jimma University for providing laboratory facility.
*Corresponding author. E-mail address: tesfaye.refera@ju.edu.et
Received 22 May 2013; accepted 20 January 2014

