










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.30 no.5 Coimbra set. 2012
https://doi.org/10.4152/pea.201205335
Comparative Study of Different Plasticized Membrane Electrodes for the Stability-indicative Determination of Torsemide
H.E. Zaazaa, S.S. Abbas, Hebat A.M. Essam* and M. G. El-Bardicy
Analytical Chemistry Department, Faculty of Pharmacy-Cairo University, Kasr El-Aini Street, 11562 Cairo, Egypt
Abstract
Four torsemide-selective electrodes all on solid state graphite support were developed and characterized in polyvinylchloride matrices. Precipitation-based technique with tetraphenylborate (TPB) as an electroactive material in polyvinylchloride (PVC) matrix was used for sensor 1 fabrication without incorporation of an ionophore. 2hydroxypropyl- β-cyclodextrin (HP-β-CD) based technique with tetraphenylborate and either dioctyl phatalate (DOP), dibutyl sebasate (DBS) or 2-nitrophenyl octyl ether (o- NPOE) as plastisizer in carboxylated polyvinylchloride (PVC-COOH) matrix were used for sensor 2, 3 and 4 fabrications, respectively. Fast and stable Nernstian responses were evaluated according to IUPAC recommendations in the concentration ranges from 1 × 10-5 to 1 × 10-3 M for sensor 1 , from 1 × 10-5 to 1 × 10-2 M for sensor 2, from 1 × 10-6 to 1 × 10-4 M for sensor 3, and from 1 × 10-6 to 1 × 10-3 M for sensor 4. The sensors show good selectivity to the drug in presence of a variety of inorganic and organic interferents including acid degradation product of torsemide, related substances and pharmaceutical excipients. Validation of the method showed the suitability of the proposed electrodes for the use in the quality control assessment of the drug.
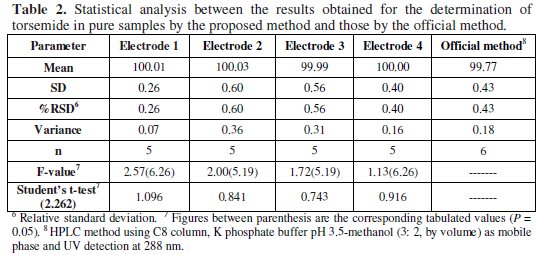
Furthermore, statistical comparison between the results obtained by the proposed method and the official method of the drug was performed and no significant difference was found.
Keywords: Torsemide-selective electrode; graphite electrode; 2-hydroxy propyl-βcyclodextrin; stability indicating study; pharmaceutical dosage form.
Introduction
Torsemide (TOR) is a sulfonylurea derivative, used as a loop diuretic, for treatment of oedema associated with heart failure, renal and hepatic disorders, also used in the treatment of hypertension, either alone or with other antihypertensives [1]. Chemically it is N-[{(1 methylethyl) amino} carbonyl]-4[( 3-methylphenyl) amino]-3-pyridinesulfonamide [2]. Torsemide was determined by pharmacopeial and non pharmacopeial methods where it is assayed in the British pharmacopoeia via non-aqueous titration [3], while assayed in the USP pharmacopeia by HPLC method [4]. The non pharmacopeial methods used for determination of TOR include HPLC [5-15], LC/MS [6-21], GC/MS [22, 23], CZE [24], spectrophotometry [25-29], multivariate optimization of CZE [30], voltammetric determination using carbon past electrode [31] and dropping Hg electrode [32] and TLC-densitometry [33]. Stability-indicating HPTLC [34] and HPLC [35] methods for analysis of TOR has been recently reported.
Modern techniques based on material transport across a specific membrane as ion selective electrodes (ISEs) are now widely used in the determination of trace amounts of analytes as well as drugs in pure form and pharmaceutical dosage forms [36]. The material transport includes both neutral and charged complex species, and simple ions [37, 38]. The high selectivity of these electrodes imparts a great advantage over other techniques [39]. Analytes in colored, turbid or viscous samples can be determined accurately. They show rapid responses to changes in the concentration. Furthermore, they may be used for measurement over a wide concentration range. ISEs are generally tolerant of small pH changes. A further advantage is that they are relatively cheap and simple to develop, set up and run with no need for sophisticated apparatus compared with other published TLC or HPLC methods. Moreover, the chemical design of the electrodes has been developed to give superior selectivity and response [40]. The scientific novelty of this work is the use of ISEs which has several advantages for the determination of TOR in the presence of its degradation products in powder form, laboratory prepared mixtures and in pharmaceutical formulation.
Experimental
Instruments
Jenway digital ion analyzer model 3330 (UK) with Ag/AgCl double junction reference electrode No. Z113107-1EAPW (Aldrich Chemical Co.) was used. The influence of pH on the response of the electrodes was studied using a pH glass electrode Jenway (Jenway, UK) No. 924005-BO3-Q11C. The determination of the samples occurred using a Magnetic stirrer, Bandelin Sonorox, Rx510S (Budapest,Hungaria).
Materials and reagents
Pure standard
Standard TOR was kindly supplied by Multi-Apex pharma, Badr City, Cairo, Egypt. It was assayed for its purity according to the USP method [4] and found to contain 99.77%.
Pharmaceutical formulation
Examide® tablets, manufactured by Multi-Apex pharma, Badr City, Cairo, Egypt. Batch No. 1230311 & 1240311, labelled to contain 10 & 20 mg of torsemide/ tablet, respectively.
Degraded sample
Accelerated acid-degradation was performed by dissolving 25 mg of pure TOR powder in 25 mL of 2 N hydrochloric acid, then the solution was refluxed for 4 hrs. The solution was neutralized to pH 7.0 by 2 N sodium hydroxide and complete degradation was followed by TLC using acetone-chloroform-ethyl acetate (40: 40: 20, by volume) as developing system. The neutralized solution was evaporated under vacuum nearly to dryness, then the degradation product was re-crystallized from methanol. The obtained degradation product was elucidated and confirmed by IR and mass spectrometry.
Chemicals and reagents
All chemicals and solvents used were of analytical grade (double-distilled water was used).
Polyvinylchloride (PVC), high molecular weight, polyvinylchloride carboxylated (PVC-COOH) and 2-hydroxypropyl-β-cyclodextrin (HP-β-CD) were purchased from Fluka chemie (GmbH Germany). Tetrahydrofuran (THF) was purchased from BDH (limited Poole, England), while 2-nitrophenyl octyl ether (o-NPOE), dioctyl phthalate (DOP), dibutyl sebasate (DBS) and tetraphenylborate (TPB) were from Sigma/Aldrich (St.Louis, MO). Sodium hydroxide, 2 M aqueous solution and hydrochloric acid, 2 M aqueous solution, were prepared and obtained from Prolabo (VWR International, West Chester, PA). Also, Britton- Robinson buffer (BRB) (pH 2-11) [41] was prepared by mixing different volumes of 0.04 M acetic acid, 0.04 M phosphoric acid, 0.04 M boric acid and 0.2 M sodium hydroxide.
Standard solutions
(a) Torsemide stock solution (1 × 10-1 M). It was freshly prepared daily by transferring 3.48 g of TOR into a 100-mL volumetric flask, then dissolving in 90-mL double-distilled water; 0.4-mL of concentrated HCl were added till clear solution and the volume was completed with water. The stability of the prepared solution was studied, and it has been found to be stable with no apparent degradation at least for 24 h at 25 °C.
(b) Torsemide working solutions (1 × 10-6 to 1 × 10-2 M). It was freshly prepared by suitable dilution from its stock solution using BRB pH 5.
(c) Working standard solution of the acid degradation products (1 × 10-2 M) derived from complete degradation of 10-mL of (1 × 10-1 M) standard solution of TOR in 2 N HCl. The degraded solution was neutralised, then transferred quantitatively into a 100 mL volumetric flask and completed to volume with BRB pH 5.
Procedures
Fabrication of membrane sensors
(a) Precipitation-based technique for the preparation of PVC-membrane sensor (sensor 1)
Ten milliliters of 1 × 10-2 M aqueous solution of TOR were mixed with 10 mL of a saturated aqueous solution of 1 × 10-2 TPB solution. The resulting precipitate was filtered using Whatman (Florham Park, NJ) No. 42 paper, washed with cold water, allowed to dry at room temperature and grounded to fine powder, forming the ion-pair association complex where elemental analysis of the formed complex was performed. In a glass Petri dish (5 cm diameter), 10 mg of the previous association complex were mixed with 0.4 mL of DOP, then 0.19 gm of PVC were added. The mixture was dissolved in 5 mL THF, and then the Petri dish was covered with a filter paper and left to stand for one hour to allow slow evaporation of the solvent, producing a thick homogeneous master coating PVC solution.
A rod of spectrographic graphite (5 mm in diameter and 15 mm in length) was inserted in a polyethylene sleeve, and about 3 mm of the other end of the protruded rod served as a measuring surface. This end of the rod was washed with acetone, dried in air for 3 hrs, and dipped rapidly into the previously prepared PVC solution.
The solvent was allowed to evaporate in air after each dipping, and the dipping process was repeated 6-8 times to produce a uniform membrane on the surface of the graphite rod. One drop of mercury was added in the polyethylene sleeve to ensure electrical contact with the connection cable. The coated graphite rod was conditioned by soaking in a 1 × 10-2 M TOR solution for 2 hrs, and stored in the same solution when not in use.
(b) Hydroxypropyl-β-cyclodextrin-based technique for the preparation of PVCCOOH- membrane sensors (sensors 2, 3 and 4)
In a glass Petri dish (5 cm diameter), 10 mg of TPB and 0.04 g of HP-β-CD were mixed thoroughly with 0.4 mL of DOP, DBS or o-NPOE and 0.19 g PVC-COOH for the preparation of sensors 2, 3 and 4, respectively. The mixture was dissolved in 5 mL THF, and then the Petri dish was covered with a filter paper and left to stand for one hour to allow slow evaporation of the solvent, producing a thick homogeneous master coating PVC-COOH solution, and the procedure was completed as under the same conditions of sensor 1.
Sensors calibration
The conditioned sensors were calibrated by separately transferring 50 mL aliquots of solution covering the concentration range of 1×10-6 to 1×10-2 M drug into a series of 100-mL beakers; the electrode system was immersed in each solution in conjunction with an Ag/AgCl reference electrode. The emf within ±1 mV readings were recorded after equilibrate while stirring. The membrane sensor was stored in deionized bidistilled water between measurements; the electrode potential was plotted versus each negative logarithmic concentration of drug. The obtained calibration plot was used for subsequent measurements of unknown samples of TOR.
Effect of pH
The effect of pH on the response of the investigated electrodes was studied using 1 × 10-4 and 1 × 10-5 M solutions of TOR in BRB with pH ranging from 2 to 11.
Sensors selectivity
The potentiometric selectivity coefficients (KA.B)pot of the proposed sensors towards different substances were evaluated according to IUPAC guidelines using the separate solution method [42], by applying the following equation:
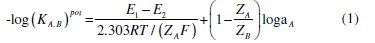
where (KA.B)pot is the potentiometric selectivity coefficient, E1 and E2 are the potential readings recorded after exposing the electrode to the same concentration of the studied drug and the interferent, respectively, ZA and ZB are the charges of TOR and interfering ion, respectively, aA is the activity of the drug and 2.303RT/ZAF represents the slope of the investigated sensors (mV/concentration decade).
Determination of torsemide in pharmaceutical formulation (Examide® tablet)
Twenty tablets of each Examide® 10 & 20 mg were weighed and finely powdered in a small dish. Amounts of the powdered tablets equivalent to 1.74 mg TOR, were accurately transferred into two 50-mL measuring flasks and the volumes were completed to the mark with BRB pH 5 to prepare a 1 × 10-4 M aqueous solution of TOR. The potential readings produced by immersing the prepared electrodes in conjunction with the double junction Ag/AgCl reference electrode in the prepared solution were recorded and compared with the calibration graphs.
Determination of torsemide in the presence of its acid degradation product
In a series of 100-mL volumetric flasks, different aliquots (1, 2, 3, 4, 5, … 9 mL) of TOR solution (1 × 10-4 M) were quantitatively and separately transferred. Complementary aliquots from the corresponding degradation product solution (1 × 10-4 M) were added, to prepare mixtures containing 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9 TOR: degradation product, respectively. The emf values of these laboratory-prepared mixtures were recorded and the results were compared with the calibration plot.
Results and discussion
The stability of TOR was studied according to ICH guidelines Q1A (R2) [43] for:
(a) Stress acid and alkaline: 1 M HCl/1 M NaOH for 5 hrs, 2 M HCl/2 M NaOH for 4 hrs.
(b) Oxidative condition: 3% H2O2 for 2 hrs, 4 hrs and 6 hrs. The degradation process under the previously mentioned conditions was followed using TLC and the compound was found to be liable to degradation under acidic, basic and oxidative conditions giving the same two degradation products; one of them (deg 1) is among the impurities stated in USP, while the other degradation product is an aliphatic fragment with no UV absorption (deg 2). Acid stress condition was the stress condition of choice for preparing the degradation products as it provides the least drastic condition, (Fig. 1).
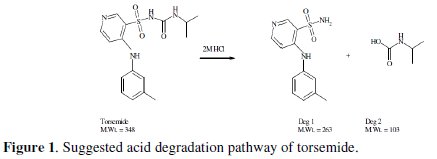
Since this work was concerned with the development of stability-indicating methods for the determination of TOR, the degradation products were prepared in laboratory, as mentioned in the section of degraded sample. The structure of the isolated acid degradation product was confirmed using IR and MS spectroscopy, (Fig. 2a-c).
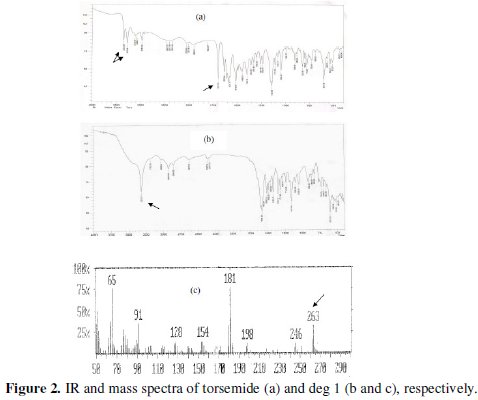
The assignment of TOR degradation product was based on comparison of IR spectral data for the separated compound with that of the intact drug. The IR spectrum of the degradation product showed disappearance of NH-amide peak at 3278.99 cm-1 and C=O-amide at 1697 cm-1 in the parent compound and appearance of a new sharp and intense peak of -NH-and -NH2 groups at 3313 cm-1, which indicates the breaking of amide linkage, (Fig. 2 a & b). Moreover, the mass spectrum of deg1 showed a molecular ion peak at m/z 263 (Fig. 2c). This finding suggests the degradation pathway and indicates the structure of the degradation product of TOR, as illustrated in (Fig. 1). The inclusion complexation and molecular recognition are of current interest in host-guest and supramolecular chemistry and offer a promising approach to chemical sensing [44, 45]. The use of selective inclusion complexation and complementary ionic or hydrogen bonding are two main strategies for preparing synthetic host molecules, which recognise the structure of guest molecules [46]. Natural or synthetic modified cyclodextrins (CD) are viewed as molecular receptors. In the case of natural CD, cooperative binding with certain guest molecules was mostly attributed to intermolecular hydrogen bonding between the CD molecules, while intermolecular interactions between the host and guest molecules (hydrogen bonds, hydrophobic interactions and Van der Waals forces) contributed to cooperative binding processes when synthetic CDs were used [47]. Although the size and geometry of the guest mainly govern the binding strength, it is possible to modifying the host-guest interactions through the three hydroxyl groups on each glucose unit. Indeed, the use of 2-hydroxypropyl-β-cyclodextrin enhanced the interaction properties between host and guest molecules [48].
This work originates from the fact that torsemide behaves as cation, due to the presence of the secondary amine functional group. This fact suggests the use of anionic type of ion exchangers, forming water insoluble ion association complexes. Tetraphenylborate as an anionic exchanger was used for construction of water insoluble ion-association complex with TOR(sensor 1), while sensors 24 utilized hydroxypropyl-β-cyclodextrin -based technique using graphite microelectrode in all sensors, which are characterized by their small physical size that allows exploration of microscopic domains, such as biological systems, their fast response time, due to the reduced diffusion layer, allows rapid scan rates to be used and their low susceptibility to ohmic loss, due to the small currents produced, enables their uses in highly resistive biological media [49]. These microelectrodes behave as two interface devices, membrane/electrolyte interface and membrane/ metal interface [50]. Performance characteristics of these electrodes (sensors 2-4) reveal low detection limit, high sensitivity, good selectivity, fast response, long life span, and applicability for accurate determination of torsemide in presence of its acid degradation products and in dosage form.
The fact that TOR can behave as a cation suggests the use of anionic ion exchangers. It has been found that TPB was optimum anionic exchanger for the studied drug, because of the low solubility of its reaction product and its suitable grain size [37, 51]. Torsemide reacted with TPB to form a water insoluble 1:2 drug-ion association complex, as confirmed by elemental analysis. This indicates that the drug behaves as a divalent species. The complex was prepared, characterized, and incorporated with a suitable solvent mediator in PVC matrix membranes.
Fabrication and performance characteristics of the sensors
It has been reported that PVC matrix is a regular support and reproducible trap for ion association complexes in ISEs. Nevertheless, its use creates a need for plasticization and places a constraint on the choice of the mediator [52]. In this study, PVC was used in the fabrication of sensor 1, while PVC-COOH with HP β-CD and different plasticizers were used in fabrication of the other proposed sensors. Cyclodextrins are optically active oligosaccharides that form inclusion compounds in aqueous and in solid states with organic molecules. HP-β-CDbased sensors showed accurate results in both response and selectivity. Also, the improved stability and selectivity are attributed to the increase in the acidity of the carboxylate group of PVC-COOH and hence, interacting with the drug.
Plasticizers play an important role in the behaviour of polymeric membrane ion selective electrodes. For a plasticizer to be adequate for its use in polymeric membrane ion selective electrodes, it should gather certain properties and characteristics such as having high lipophilicity, high molecular weight, and low tendency for exudation from the polymeric matrix [53]. The introduction of polar or polarisable groups into membranes reduces the rate of exudation and therefore the loss of membrane components by their migration to the aqueous phase which reduces the selectivity of membranes. Additionally, plasticizer viscosity and dielectric constant should be adequate.
It is well known that the sensitivity and selectivity of ion-selective electrodes strongly depend on the membrane compositions and the nature of the plasticizer used [54-56]. The nature of the plasticizer influences the dielectric constant of the membrane phase, the mobility of the ionophore molecules, and the forms of the ligands [57, 58]. To investigate the effect of plasticizers, PVC-COOH membranes with different plasticizers DOP, DBS and o-NPOE were prepared using TPB as the sensing membrane components. The electrochemical performance characteristics of the sensors were systematically evaluated according to IUPAC recommendations [42]; the potentiometric responses and results including slope, response time and working concentration range are summarized in Table 1.
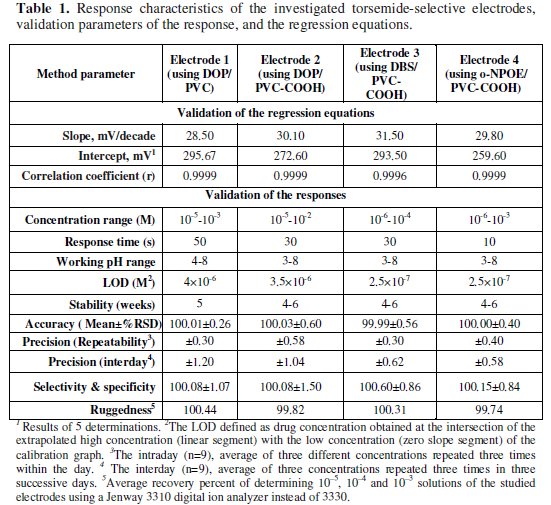
According to the data presented in Table 1, o-NPOE is the most effective of the three different plasticizers used in sensors 2-4. This indicates that o-NPOE plasticizes the membrane and adjusts both the membrane permittivity and the mobility of the ion-exchanger sites to facilitate the inclusion of organic molecules by competitive inclusion and give the optimal selectivity and sensitivity [59].
The electrochemical cell of the suggested membrane electrodes for the determination of TOR can be illustrated diagrammatically as follows:

Calibration and dynamic response time of sensors
The potential displayed by the 4 proposed electrodes for constructive measurements of standard drug solutions in the same day and from day-to-day did not vary by more than ±1 mV. Calibration slopes did not change by more than ±2 mV/decade concentration over a period of 4-6 weeks for electrodes 1-4, Table 1. The dynamic response times for the electrodes to reach values within ± 1 mV of the final equilibrium potential after increasing the drug concentration 10-fold were found to be 50, 30, 30 and 10 s for electrodes 1, 2, 3 and 4, respectively, Table 1. The slopes of the calibration graphs were 28.5, 30.10, 31.50, and 29.80 mV/concentration decade for electrodes 1, 2, 3 and 4, respectively. These are typical values of divalent substances; torsemide behaves as divalent cation via its 2 amino groups. Fig. 3 shows a decrease in the negative potential as the concentration increases due to the decrease in the negative charge on the membrane.
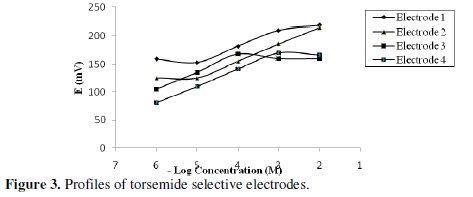
Deviation from the ideal Nernstian slope (30 mV/decade), stems from the fact that the electrodes respond to the activity of the drug rather than its concentration. The investigated electrodes exhibit fast response time (10 - 50 s) and fair stability (4 - 6 weeks). The fabricated ISEs gave a Nernstian response within a 1 × 10-6 - 1 × 10-2 M concentration range. The proposed method was compared with the USP HPLC method [4] and no significant difference was observed, as seen in Table 2.
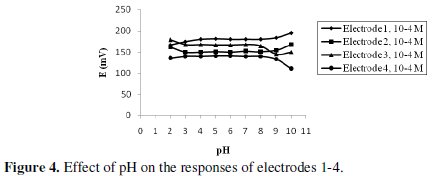
Temperature and pH of the sensors
For quantitative measurements with ISEs, studies were performed to reach the optimum experimental conditions. The pH effect was optimized from the point of view of both sensor function and chemical form of the test substances. It is apparent that the investigated electrodes gave a useful pH range from 3-8. Above and below this range, the potential displayed by the electrodes was noisy. Above pH 8, the potential showed a sharp decrease due to the formation of nonprotonated amino group of TOR. Below pH 3, the membranes may extract H+, leading to noisy responses, Fig. 4.
The effect of temperature was also studied by monitoring the potential response displayed by the electrodes as a function of -log of the drug concentration at 20, 30, and 35 °C. It was found that the suggested electrodes exhibited a gradual increase in their potentials as the temperature increased in the range of 20 - 35 °C; however, the calibration graphs obtained at different temperatures were parallel. In spite of this, the limit of detection and response time did not significantly vary with changes of temperature, indicating reasonable thermal stability of TOR membranes up to 35 °C.
Selectivity of sensors
The influence of various basic substances as amino compounds of biological interest (urea, glycine, and hydroxylamine) and other inorganic cations (K+, Na+, NH4+, Mg2+, and Ca2+) that are usually found in biological fluids on the response of sensors was investigated by measuring the potentiometric influence of some potentially interfering speci es. The selectivity coefficients were calculated by the separate solution method [60], in which potentials were measured for 1 × 10-4 M aqueous drug solution, and then for 1 × 10-4 M aqueous interferent solution, separately. The results in Table 3 reveal that all investigated sensors display high selectivity, but sensors 2 - 4 are at least 10 - 100 times more selective than sensor 1.
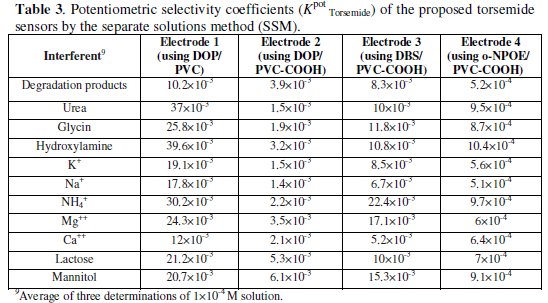
Moreover, sensor 4 displays higher selectivity and lower response for the potentially interfering species than sensors 2 & 3. Table 4 shows the results obtained upon analysis of synthetic mixtures containing different ratios of intact drug and degraded samples varying from 10:90 to 90:10.
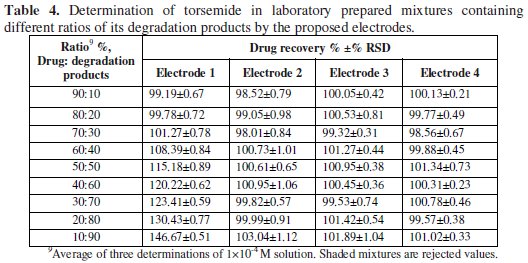
The results show that sensors 2 - 4 can be successfully used for selective determination of intact drug in the presence of up to 80% of its degradation products. Sensor 1 suffers from high interference when the degradation products concentration reaches about 30%. Thus, sensors 2 - 4 are recommended for use in stability-indicating methods. This can be attributed to the preferential interaction between the TOR cation and very polar OH-groups present in the HP-β-CD structure and the acidic group in PVC-COOH in presence of a plasticizer with high dielectric constant, as o-NPOE. Table 5 shows the results obtained for the determination of TOR in its pharmaceutical formulations, proving the applicability of the method.
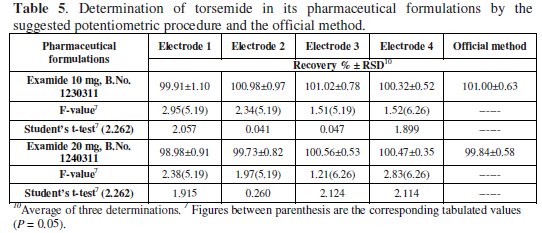
Linearity was assessed by the determination of the same concentration range as the calibration graph. The precision and mean accuracies are given in Table 1. To evaluate precision, 3 concentrations within the linear range (1 × 10-5, 1 × 10-4, and 1 × 10-3 M solutions of TOR) were chosen. Three solutions of each concentration were prepared and analyzed in triplicate (repeatability assay). This assay was repeated on 3 different days (intermediate assay). To study the method ruggedness, 1 × 10-5, 1 × 10-4, and 1 × 10-3 M solutions of TOR were analyzed by the 4 studied electrodes using a Jenway 3310 digital ion analyzer instead of the Model 3330; the results were stable upon change of the instrument. In summary, changes of variables and apparatus did not affect the accuracy of the results (Table 1).
Validation of the proposed potentiometric method for determining TOR drug was made by measuring range, limit of detection (LOD), accuracy, precision, repeatability, intermediate precision, linearity, sensitivity, and ruggedness. Results obtained are depicted in Table 1. These data indicate that the proposed method is applicable for the quality control of the drug formulations.
Conclusions
The described sensors are sufficiently simple and selective for the quantitative determination of TOR in pure form, pharmaceutical formulations, and in the presence of its degradation products. The utility of PVC-COOH as polymer has a significant influence on increasing both membrane sensitivity and selectivity of sensors 2, 3 and 4. Electrode 4 was faster than the others, thus the response time was more or less instantaneous (10 min for electrode 4), while those of 1, 2 and 3 were about 0.5-1 min. The precision of sensors 2 and 4 was higher than that of sensors 1 and 3 (less than one). The use of the proposed sensors offers advantages over chromatographic techniques, providing fast response, elimination of drug pretreatment and separation steps, lacking of expensive and sophisticated apparatus; furthermore, there is no need of expensive and special grade solvents. They can therefore, be used for routine analysis of TOR in quality control laboratories.
References
1. Sweetman SC, editor. Martindale The Complete Drug Reference. 36th ed. London, UK: Pharmaceutical Press; 2009. [ Links ]
2. Moffat AC, Osselton MD, Widdop B. Clarke's Analysis of Drugs and Poisons in Pharmaceuticals, Body Fluids and Post-mortem Materials. 3rd ed. London: Pharmaceutical Press; 2004. P. 1652. [ Links ]
3. British Pharmacopoeia. London, UK: The Stationery Office on Behalf of the MHRA; 2010. II:2124-2125. [ Links ]
4. The U.S. Pharmacopeia. 30th ed. Rockville, MD: US Pharmacopeial Convention; 2007. P. 3385-3386. [ Links ]
5. Barroso MB, Alonso RM, R.M. Jimenez RM. J Liq Chromatogr Related Technol. 1996;19:179-186.
6. Barroso MB, Alonso RM, Jimenez RM. J Chromatogr Sci. 2001;39:491496. [ Links ]
7. Qin Y, Zhu ST, Wang C, Zhao M, Peng SQ. Fenxi Ceshi Xuebao. 2002;21:85-87.
8. Engelhardt S, Meineke I, Brockmoeller J. J Chromatogr B: Anal Technol Biomed Life Sci. 2006;831:31-35. [ Links ]
9. Liu KH, Lee YK, Ryu JY, Lee DJ, Kang W, Lee SS, Yoon YR, Shin JG. Chromatographia. 2004;60:639-643.
10. Qin Y, Wang XB, Wang C, Zhao M, Wu MT, Xu YX, Peng SQ. J Chromatogr B: Anal Technol Biomed Life Sci. 2003;794:193-203.
11. Qin Y, Zhu ST, Wang C, Ming ZO, Peng SQ. Fenxi Ceshi Xuebao. 2003;22:41-44.
12. Thieme D, Grosse J, Lang R, Mueller RK, Wahl A. J Chromatogr B: Biomed App. 2001;757:49-57.
13. Hu M, Liu YA, Du AZ. Yaowu Fenxi Zazhi. 2001;21:197-198. [ Links ]
14. Karnes HT, Farthing D, Besenfelder E. J Liq Chromatogr Related Technol. 1989;12:1809-1818. [ Links ]
15. Besenfelder E. J Pharm Biomed Anal. 1987;5:259-266. [ Links ]
16. Deventer K, Van Eenoo P, Delbeke FT. Rapid Comm Mass Spect. 2005;19:90-98. [ Links ]
17. March C, Farthing D, Wells B, Besenfelder E, Karnes HT. J Pharm Sci. 1990;79:453-457.
18. Carda-Broch S, Torres-Lapasio JR, Esteve-Romero JS, Garcia-Alvarez-Coque MC, J Chromatogr. 2000;893:321-337. [ Links ]
19. Deventer K, Delbeke FT, Roels K, Van Eenoo P. Biomed Chromatogr. 2002;16:529-535. [ Links ]
20. Kolmonen M, Leinonen A, Pelander A, Ojanpera I. Anal Chim Acta. 2007;585:94-102. [ Links ]
21. Goebel C, Trout GJ, Kazlauskas R. Anal Chim Acta. 2004;502:65-74. [ Links ]
22. Barroso MB, Meiring HD, deJong A, Alonso RM, Jimenez RM. J Chromatogr B: Biomed App. 1997;690:105-113.
23. Beyer J, Bierl A, Peters FT, Maurer HH. Therapeutic Drug Monit. 2005;27:509-520. [ Links ]
24. Akesolo U, Gonzalez L, Jimenez RM, Alonso RM. Electrophoresis. 2002;23:230-236. [ Links ]
25. Shashikant BB, Sanjay KP. Int J Pharm Quality Assurance. 2010;2:52-55. [ Links ]
26. Marothu V, Dannana G, E-J Chem. 2008;5:473-478.
27. Jatin G, Gaurav K, Vishal Y, Wakode SR. J Chem Pharm Res. 2010;2:513-517. [ Links ]
28. Sankar DG, Babu PJ, Sujatha N, Latha PVM, Asian J Chem. 2006;18:3247-3248. [ Links ]
29. Krishna MV, Sankar DG. Acta Ciencia Indica Chem. 2006;32:355-358. [ Links ]
30. Akesolo U, Gonzalez LJimenez RM, Alonso RM. J Chromatogr. 2003;990:271-279. [ Links ]
31. Barroso MB, Alonso RM, Jimenez RM. The Analyst .1994;119:2359-2362. [ Links ]
32. Fernandez M, Alonso RM, Jimenez RM, Legorburu MJ. The Analyst. 1994;119:319-322. [ Links ]
33. Sharma MC, Sharmaa S, Kohlib DV, Sharmac AD. Der Pharma Chemica. 2010;2:121-126. [ Links ]
34. Kakde R, Chaudhary N, Barsagade A, Kale D. Acta Chromatographia. 2011;23:145-155. [ Links ]
35. Patel HB, Mohan A, Joshi HS, J AOAC Int. 2011;94:143-149. [ Links ]
36. Metwally FH. Yakugaku Zasshi. 2007;8:1267-1273. [ Links ]
37. Elsaharty Y, Metwally FH, El-Khateeb SZR. J Pharm Biomed Anal. 2006;41:720-724. [ Links ]
38. Mostafa GA, Al-Majed A. J Pharm Biomed Anal. 2008;48:57-61. [ Links ]
39. Conway BA. Ion Selective Electrodes. 1995;3:41. [ Links ]
40. Hassan SS, Mahmoud WH, Othman AM. Talanta. 1997;44:1087. [ Links ]
41. Vogel's Text Book of Quantitative Inorganic Analysis including elementary instrumental analysis. 4th ed. ELBS; 1978. [ Links ]
42. IUPAC Analytical Chemistry Division -Commission on Analytical Nomenclature. Pure Appl Chem. 2000;72:1851. [ Links ]
43. ICH, Q1A (R2), Stability Testing of New Drug Substances and Products, International Conference on Harmonization, IFPMA. Geneva, Switzerland; 2003. [ Links ]
44. Mittal SK, Kumar A, Gupta N, Kaur S, Kuma S. Anal Chim Acta. 2007;585:161.
45. Zanganeh AR, Amini MK. Sens Actuators B Chem. 2008;135:358. [ Links ]
46. Gorski L, Matusevich A, Parzuchowski P, Luciuk I, Malinowska E. Anal Chim Acta. 2010;665:39.
47. Sideris EE. Eur J Pharm Sci. 1999;7:271. [ Links ]
48. Flink S, van Veggel FCJM, Reinhoudt DN. Adv Mater. 2000;12:1315. [ Links ]
49. Moody G, Thomas J. Selective Ion Sensitive Electrode. Merrow Technical Library; 1979. Chap 1. [ Links ]
50. James H, Carmack G, Freiser H. Anal Chem. 1972;44:856-861. [ Links ]
51. Hassan SSM, Abou-Sekkina MM, El-Ries MA, Wassel AA. J Pharm Biomed Anal. 2003;32:175-180. [ Links ]
52. Ber A, Moody G, Thomas J. The Analyst. 1976;101:179. [ Links ]
53. Eugster R, Rosatzin T, Rusterholz B, Aebersold B, Pedrazza U, Ruegg D, Schmid A, Spichinger UA, Simon W. Anal Chim Acta. 1994;289:1-13.
54. Bakker E, Buhlmann P, Pretsch E. Electroanalysis. 1999;11:915. [ Links ]
55. Ammann E, Pretsch P, Simon W, Lindner E, Bezegh A, Pungor E. Anal Chim Acta. 1991;171:1380.
56. Bakker E, Buhlmann P, Pretsch E. Chem Rev. 1997;97:3083. [ Links ]
57. Yang X, Kumar N, Chi H, Hibbert DD, Alexander PNW. Electroanalysis. 1997;9:549.
58. Ganjali MR, Norouzi P, Alizadeh T, Adib M. Anal Chim Acta. 2006;576:275. [ Links ]
59. Gupta VK, Singh AK, Mehtab S, Gupta B. Anal Chim Acta. 2006;566:5. [ Links ]
60. Ma TS, Hassan SSM. Organic Analysis Using Ion Selective Electrodes. London, UK: Academic Press; 1982. vols 1 and 2. [ Links ]
Acknowledgements
The authors are thankful to Dr. Mohamed K. Abd El-Rahman, lecturer at the Analytical Chemistry Department, Faculty of Pharmacy, Cairo University, Egypt, for his helpful, effective and supportive instructions.
*Corresponding author. E-mail address: Heba_essam80@hotmail.com
Received 2 June 2012; accepted 31 October 2012

