










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.30 no.2 Coimbra mar. 2012
https://doi.org/10.4152/pea.201202111
Characteristics of the spontaneous deposition of Sn in polycrystalline Pt
Javier E. Rodríguez Yáñez*
Laboratorio de Química Marina, Universidad Nacional, Heredia, Costa Rica
doi: 10.4152/pea.201202111
Abstract
Spontaneous deposition of tin (Sn) in polycrystalline platinum (Pt pc) from a sulfuric acid solution of tin salts, was evaluated by conventional electrochemical techniques, to determine its characteristics. Tin was deposited from a solution of SnCl2 or SnSO4 in sulfuric acid 1 M. The degree of surface coating by Sn was calculated by evaluating the region of hydrogen electrosorption through voltammograms performed using solutions containing only the supporting electrolyte, and the supporting electrolyte containing the Sn salt, respectively. The adsorption behavior of tin and oxidation have been studied by the results obtained with platinum electrodes and electrodes of platinum/tin, considering as comparative parameter the adsorption efficiency with respect to the area free of platinum.
Keywords: tin, platinum, spontaneous, electrocatalysis, voltammetry.
Introducción
La modificación de superficies metálicas por deposición de otros metales es una de las técnicas más usuales para la generación y evaluación de nuevos catalizadores.
La química verde plantea actualmente una tendencia a la minimización de las energías necesarias para la generación de un nuevo producto, así como la disminución de la toxicidad en su producción. En este sentido, las técnicas electroquímicas son una de las metodologías empleadas, las cuales permiten optimizar la generación de estas superficies metálicas compuestas [1].
Dentro de las técnicas más usuales de obtención de estas superficies por vía electroquímica se encuentra i) el depósito de metales a potenciales positivos con respecto al potencial de la cupla metálica E0Mz+/M, llamado depósito a subpotenciales (under potential deposition o upd); ii) la deposición a potenciales más negativos que el de esa cupla metálica o deposición a sobrepotenciales (over potential deposition u opd); iii) técnicas mixtas que combinan ambos potenciales en sistemas cíclicos, derivados del concepto de estabilización dinámica, u otras técnicas complejas [2-8].
Hace algunos años surgió con algunos autores el concepto de deposición espontánea, esto es, sin aplicación de corriente o potencial. En este sistema la deposición depende de las condiciones de la superficie, del medio electrolítico y de la naturaleza del metal a depositar [2-5].
La existencia de diferentes métodos de deposición, hace particularmente atractiva la consideración de la deposición espontánea (esp), en virtud de su facilidad, rapidez y relativa estabilidad [9-14].
Dependiendo de la técnica, se logran en general cubrimientos superficiales parciales, con estabilidad y reactividad variable y compleja [15-18].
Otra consideración según la situación específica, es la relativa selectividad por algunos tipos de planos cristalinos en la deposición, según el metal a depositar y el metal base, lo cual condiciona sus posibles capacidades catalíticas [19-22].
En el caso del Sn las características del depósito formado en las distintas técnicas son discutidas ampliamente, siendo en general descriptas para el estaño como una mezcla de Sn, SnO, SnO2, Sn(OH)2, principalmente [23-38].
En general, se reporta que los depósitos de Sn se producen en islas sobre estructuras cristalinas de Pt con orientaciones (110), proponiendo que su actividad está asociada a la situación de carga parcial o a la posibilidad de dismutar desde su estado como Sn2+ [8,39,40].
Metodología
Electrodos
Los experimentos fueron realizados usando alambres de platino policristalino (pc) de 0,5 mm de diámetro y 99,999% de pureza (Good Fellow Co.) como electrodos de trabajo.
Como contra electrodo se utilizó un electrodo de platino de gran área, más de 10 cm2 y como electrodo de referencia un electrodo normal de hidrógeno (ENH) generado con la disolución de trabajo. Todas las medidas se refieren al ENH.
El área real del electrodo de trabajo fue determinada por integración matemática del área de adsorción de hidrógeno. Los resultados son expresados en función de esta área disponible.
Disoluciones
El electrolito soporte utilizado fue H2SO4 1 mol L-1 , preparado con un ácido Baker (97,8% ACS), diluido con agua de calidad Millipore MilliQ plus (resistividad 18,2 MΩ cm).
La disolución para la deposición 0,1 mmol L-1 en estaño, fue preparada a partir de SnSO4 (Sigma Aldrich 99,5%) o SnCl2.2H2O (Sigma Aldrich 99,9%) en ácido sulfúrico 1 mol L-1.
Métodos de deposición
A fin de evaluar los efectos del Sn en Pt pc, se realizaron las deposiciones del mismo, como se indica en la literatura [10,11,13,40], según la siguiente metodología:
Deposición espontánea de estaño por inmersión del platino en una disolución del mismo a circuito abierto o potencial libre de disolución. Todas las deposiciones fueron realizadas desoxigenando las disoluciones con nitrógeno de alta pureza (Air Lyquide 5.0).
Los depósitos de estaño obtenidos fueron estabilizados por voltamperometría cíclica, entre 0 V y 0,6 V, a una velocidad de barrido de 0,1 V s-1, en ácido sulfúrico 1 mol L-1.
Cálculos de cubrimiento
El valor de cubrimiento superficial (Θ) se define como [34]:
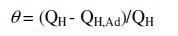
donde QH representa la carga de hidrógeno adsorbido por el electrodo de platino antes de la deposición y QH,Ad representa la carga después de la deposición, ambas en ácido sulfúrico 1 mol L-1.
Se considera para los cálculos de Q el área anódica generada entre 0,05 V y 0,4 V vs. ENH.
Para la deposición espontánea el ajuste del cubrimiento se realiza por deposiciones repetidas con estabilizaciones intermedias. Por ejemplo, para 2 minutos de deposición espontánea se obtiene alrededor de un 15 % a 20%, con una solución preparada en el día.
La disolución para deposición espontánea presenta degradación con el tiempo, decreciendo consecuentemente su efectividad, por lo cual el tiempo máximo recomendado de uso es una semana [35,41,42].
Resultados y discusión
La deposición de Sn en Pt
La superficie de platino fue modificada por deposiciones de estaño, con niveles de cubrimiento Θ del orden de 10 % a 50%. Luego de modificada la superficie se realizó su estabilización.
El perfil voltamperométrico del Pt pc a 0,1 V s-1 antes y después de la deposición de Sn puede observarse en la Fig. 1.
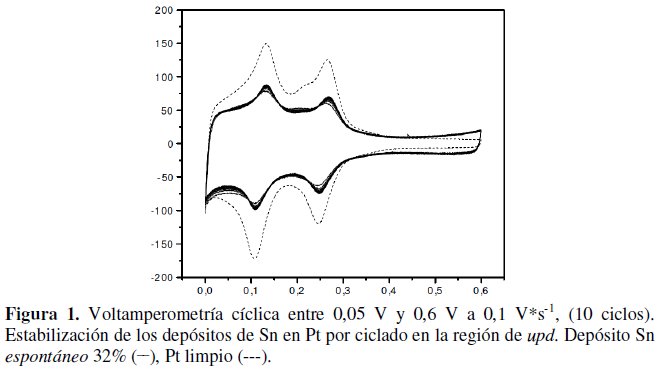
Las voltamperometrías fueron corridas entre 0,05 V y 0,6 V a fin de prevenir la desorción oxidativa del estaño depositado. Se utilizó esa región de potencial porque se ha reportado en varios artículos que el Sn depositado no se oxida pasando a disolución hasta potenciales superiores a 0,8 V [36].
La comparación entre los perfiles antes y después de la deposición evidencian la inhibición parcial de la adsorción de hidrógeno por parte del estaño depositado, así como el engrosamiento de la doble capa. Varios autores ya han indicado esta situación, marcando incluso preferencias por regiones cristalinas correspondientes al primer pico de adsorción de hidrógeno y la dependencia del efecto con el grado de cubrimiento [34,42,44].
Paralelamente se observó que se producía una estabilización de los recubrimientos generados al realizar la voltamperometria cíclica para comprobar el porcentaje de recubrimiento, generando un reordenamiento parcial de los sitios de adsorción, lo cual puede verse en la estabilización de los picos de la región de adsorción de hidrógeno en la Fig. 1.
Esta estabilización permite una mejor repetibilidad del proceso de deposición espontánea, lográndose de ese modo realizar varias deposiciones sobre la superficie, con valores de cubrimiento similares, según las condiciones de deposición utilizadas y llegando a obtener cubrimientos mayores que 50%.
Tiempo de deposición
Como primera consideración se realizó la deposición con la misma disolución, variando el tiempo de deposición y el tiempo de vida de la solución. Se observó que el tiempo de deposición es relativamente rápido, obteniendo porcentajes de cubrimientos similares y estables del orden del 15 al 20 %, para tiempos de unos 2 minutos.
En la Fig. 2, puede verse el crecimiento de la deposición espontánea con el tiempo, para una disolución de una semana.
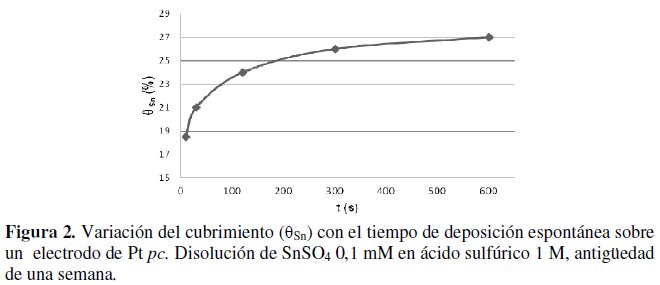
En ella se aprecia el crecimiento con el tiempo de la deposición espontánea, llegando a una meseta cerca de los 10 min. Esto plantea una relación de tipo logarítmica entre la adsorción y el tiempo de interacción, del tipo:
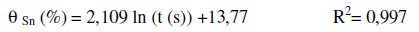
Por otro lado la edad de la disolución mostró ser una variable importante, obteniéndose una dependencia con la misma en la deposición espontánea que se producía. Los depósitos pasaban de valores de más del 30% para disoluciones recién preparadas (freshly prepared), a valores del orden de 20% para disoluciones de una semana de preparadas, con tiempos de deposición de unos 10 min, lo cual puede apreciarse en la Fig. 3 [13,23,45,46].
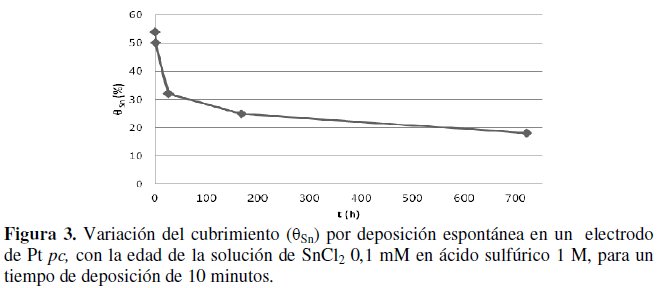
El decaimiento se puede aproximar para los valores luego de 1 h, a una curva logarítmica, según la ecuación:
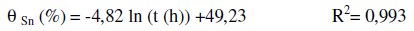
De este comportamiento y considerando una adsorción espontánea de Sn, basado en una isoterma de Frumkin del tipo:
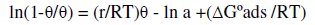
La misma presenta valores de interacción lateral de repulsión r = -14 kJ mol-1 y un ΔG0ads = -26 kJ mol-1, asimilable a una adsorción física dado que es menor que -40 kJ mol-1 [47].
Efecto del contraión
La influencia del contraión asociado al estaño, se probó midiendo los niveles de adsorción para una disolución de una semana, con SnSO4 o SnCl2.
El proceso indicó que la reacción es débilmente dependiente del contraión, debido a que en ambos casos se midieron adsorciones similares, aunque las soluciones de sulfato de estaño presentaron adsorciones ligeramente mayores, pero no significativas.
Estabilidad de la deposición espontanea
La estabilidad de los depósitos generados se visualizó inicialmente realizando voltamperometrías cíclicas en la región de potenciales de estabilidad termodinámica del Pt en ácido sulfúrico 1 M. Las mismas fueran realizadas considerando dos situaciones con el objetivo de obtener altos cubrimientos similares: i) la utilización de soluciones recién preparadas sin estabilización y ii) la utilización de deposiciones espontáneas múltiples estabilizadas. Estas situaciones se comparan con el electrodo de Pt pc y se muestran en la Fig. 4.
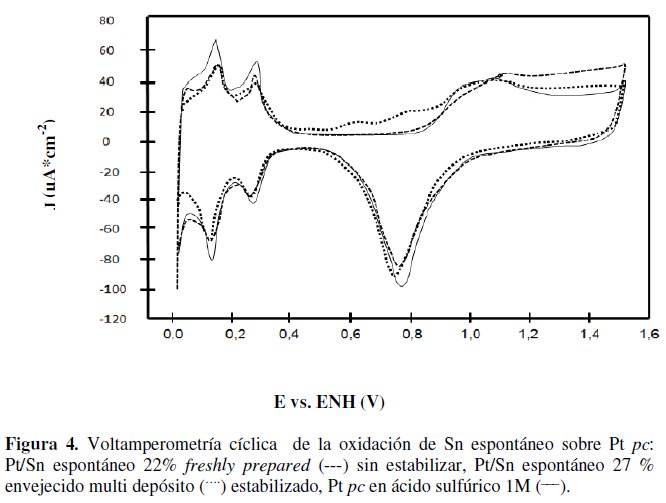
Se aprecia picos de oxidación a potenciales superiores a 0,6 V para un depósito recién preparado sin estabilización, mientras que para deposición en etapas múltiples estabilizadas, los picos son leves o mínimos, dándose una deformación de la región de óxidos de Pt [10,11,25-34]. Asimismo se realizaron cronopotenciometrías de los depósitos frente a la solución de deposición de Sn2+ 2 μM en ácido sulfúrico 1 M. Se plantearon tres situaciones básicas: i) el electrodo de Pt pc limpio; ii) un electrodo de Pt/Sn con un depósito de espontáneo sin estabilizar y iii) un electrodo de Pt/Sn con un depósito de espontáneo estabilizado, que se muestran en la Fig. 5.
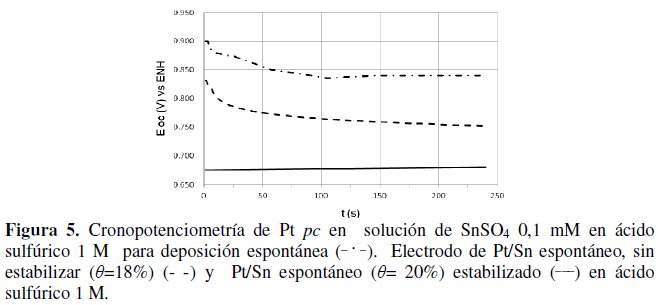
Observando los resultados se deduce que el proceso de adsorción superficial sobre Pt pc limpio tiene un potencial inicial similar al del pzc, que decae con el tiempo hasta estabilizarse a unos 100 s, alcanzando una meseta de potencial [48,49].
Por otro lado la existencia de un depósito espontáneo previo sin estabilizar, genera un decaimiento continuo del potencial. Pero cuando el mismo es estabilizado previamente, el valor de potencial es casi constante.
Discusión de resultados
Los experimentos cronopotenciométricos relacionados con la deposición indican que el proceso modifica la carga superficial, estabilizando el potencial de reposo en valores unos 50 mV menores. Esto puede asociarse a que la adsorción primaria del Sn2+ sobre la superficie de Pt pc, que de acuerdo a los resultados de la isoterma de adsorción es de tipo físico, confirmado por los valores de DGads, conduciendo a la modificación del potencial del sistema superficial. La estabilización posterior por ciclado potenciodinámico en la región de upd (0,05 a 0,6 V), permite obtener una adsorción más fuerte que la inicial, debido a que se está en una región de adsorción química. Asimismo, se reordenan los sitios de adsorción como puede observarse a través de la modificación de los picos de adsorción de H.
El ciclado potenciodinámico mejora la estabilización del depósito frente a la oxidación del mismo al aumentar el potencial y lleva el potencial de reposo a 0,6 V valores ligeramente superior al límite de la región de upd.
De acuerdo a la bibliografía, los picos de oxidación se pueden asociar a las oxidaciones sucesivas de Sn, sus óxidos o hidróxidos superficiales adsorbidos [23-38].
La existencia de depósitos previos (estabilizados o no) presenta potenciales de circuito abierto menores y una superficie modificada donde el Sn tiende más fácilmente a adsorberse.
También se puede ver que las deposiciones con bajos recubrimientos superficiales (Θ), realizados sucesivamente y estabilizados cada vez, mejoran el comportamiento final del depósito de Sn frente a la oxidación. Este hecho está posiblemente asociado a la formación de islas de Sn sobre la superficie de Pt [13,34,35].
El límite obtenido de un 60% de cubrimiento puede ser asociado a los niveles de repulsión encontrados para la adsorción física inicial. El aumento de la edad de la disolución empleada para la deposición de Sn genera depósitos más estables, no obstante, presentan menores porcentajes de recubrimiento superficial (Θ).
A concentraciones tan diluidas como las empleadas en este trabajo, el efecto del contraión no es significativo respecto al del medio.
Referencias
1. A.M. Peiro, Nuevas aportaciones al desarrollo de Metodologías en Química Verde, Tesis Doctoral, Universidad Autónoma de Barcelona, 2003. [ Links ]
2. O.A. Petrii, V.D. Kalinin, Rus. J. Electrochem. 35 (1999) 627. [ Links ]
3. M.M.P. Janssen, J. Moolhuysen, Electrochim. Acta 21 (1976) 869. [ Links ]
4. J.C. Davies, B.E. Hayden, D.J. Pegg, M.E. Rendall, Surf. Sci. 496 (2002) 110. [ Links ]
5. P. Waszczuck, J. Solla-Guillon, H.S. Kim et all, J. Catal. 203 (2001) 1.
6. J. Mc. Breen, S. Mukerjee, J. Electrochem. Soc. 142 (1995) 3399. [ Links ]
7. H. von Schenck, E. Janin, O. Tjernberg et all, Surf. Sci. 526 (2003) 184. [ Links ]
8. Yu. Morimoto, E.B. Yeager, J. Electroanal. Chem. 441 (1998) 77.
9. M. Watanabe, Y. Furuuchi and S. Motoo, J. Electroanal. Chem. 191 (1985) 367. [ Links ]
10. G. Stalnionis, L. Tamasauskaite-Tamasiunaite et all, J. Solid State Electrochem. 8 (2004) 892. [ Links ]
11. G. Stalnionis, L. Tamasauskaite-Tamasiunaite et all, J. Solid State Electrochem. 8 (2004) 900. [ Links ]
12. Y. Ishikawa, M. Liao, C. R. Cabrera, Surf. Sci. 463 (2000) 66. [ Links ]
13. S.H. Bonilla, C.F. Zinola, J. Rodríguez et all, J. Colloid. Int. Sci. 288 (2005) 377.
14. A.N. Frumkin, B.I. Podlovchenko, Ber. Akad. Wiss. USSR 150 (1963) 34. [ Links ]
15. S.J.C. Cleghorn, X. Ren, T.E. Springer et all, Int. J. Hydrogen Energy 22 (1997) 1137.
16. B. Gurau, R. Viswanathan, R. Liu et all, J. Phys. Chem. B 102 (1998) 9997.
17. H. Gerischer, D.M. Kolb, J.K. Sass, Adv. Phys. 27 (1978) 437. [ Links ]
18. K.A. Friedich, K.P.Geyzers, A.J. Dickinson, U. Stimming, Surf. Sci., 402404 (1998) 571. [ Links ]
19. C.K. Rhee, M. Wakisaka, Y.V. Tolmachev et all, J. Electroanal. Chem. 554555 (2003) 367.
20. A. Crown, A. Wieckowski, Phys. Chem. Chem. Phys. 3 (2001) 3290. [ Links ]
21. T. Frelink, W .Visscher, J.A.R. van Veen, Langmuir 12 (1996) 3702. [ Links ]
22. R. Liu, H. Iddir, Q. Fan et all, J. Phys. Chem. B 104 (2000) 3518.
23. E.V. Spinace, A.O. Neto, M. Linardi, J. Power Sources 129 (2004) 121. [ Links ]
24. F. Colom, M. González-Tejera, J. Appl. Electrochem. 24 (1994) 426. [ Links ]
25. A. Crown, I. de Moraes, A. Wieckowski, J. Electroanal. Chem. 500 (2001) 333. [ Links ]
26. F. Rodríguez, T. Morante, C.R. Cabrera, J. Electroanal. Chem. 571 (2004) 15. [ Links ]
27. Y. Li, B.E. Koel, Surf. Sci. 330 (1995) 193. [ Links ]
28. C. Gallis, B. Legrand, G. Traglia, Surf. Sci. 377-379 (1997) 1033. [ Links ]
29. M.C. Santos, L.O.S. Bulhoes, Electrochim. Acta 48 (2003) 2607. [ Links ]
30. F.P. Gorstema, J.W. Cobble, J. Am. Chem. Soc. 83 (1961) 4317. [ Links ]
31. M. Watanabe, S. Motoo, J. Electroanal. Chem. 60(1975) 267. [ Links ]
32. M. Seruga, M. Metikos-Hukovic, T. Valla et all, J. Electroanal. Chem. 407 (1996) 83.
33. R. Holze, B.B. Cattaneo, Electrochem. Acta 33 (1988) 353. [ Links ]
34. C.F. Zinola, J. Rodríguez, G. Obal, J. Appl. Electrochem. 31 (2001) 1293. [ Links ]
35. B.B. Cattaneo, T. Iwasita, J. Electroanal. Chem. 238 (1987) 151. [ Links ]
36. J. Sobkowski, K. Franaszczuk and A. Piasecki, J. Electroanal. Chem. 196 (1985) 145. [ Links ]
37. M.M.P. Janssen, J. Moolhuysen, J. Catal. 46 (1976) 289. [ Links ]
38. E. Reddington, A. Sapienza, B. Gurau et all, Science 280 (1998) 1735.
39. N.M. Markovic, P.N. Ross, Surf. Sci. Rep. 45 (2002) 117. [ Links ]
40. H. Massong, S. Tillmann, T. Langkau et all, Electrochim. Acta 44 (1998) 1379.
41. V. Solis, T. Iwasita, A. Pavese, W. Vielstich, J. Electroanal. Chem. 255 (1988) 155.
42. S. Szabo, J. Electroanal. Chem. 172 (1984) 359. [ Links ]
43. I. Bakos, S. Szabo, Electrochim. Acta 46 (2001) 2507. [ Links ]
44. X.H. Xia, Electrochim. Acta 45 (1999) 1057. [ Links ]
45. T. Iwasita, H. Hoster, A. John-Anacker et all, Langmuir 16 (2000) 522.
46. P. Waszczuck, T.M. Barnard, C. Rice et all, Electrochem. Comm. 4 (2002) 599.
47. P.W. Atkins, Química Física, Ed. Médica Panamericana, 8a edición, México, 2007. [ Links ]
48. C.M. Brett, A.M. Oliveira, Electrochemistry Principles, Methods, and Applications, Oxford University Press, New York, USA, 1993. [ Links ]
49. R. Gomez, V. Climent, J.M. Feliu, M.J. Weaver, J. Phys. Chem. B 104 (2000) 597. [ Links ]
Agradecimiento
Se agradece al Ministerio de Ciencia y Tecnología de Costa Rica, por el apoyo brindado al autor de este trabajo como becario de ese Ministerio.
*Corresponding author. E-mail address: urutico@gmail.com
Received 18 April 2012; accepted 30 April 2012

