










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta v.28 n.1 Coimbra 2010
Spectroelectrochemical Study of a Series of Fused Oligothiophenes
R. Malavé Osuna,1 C. Capel Ferrón,1 B. Vercelli,2 V. Hernández,1,* G. Zotti,2 J.T. López Navarrete1
1 Departamento de Química Física, Universidad de Málaga, 29071 Málaga,Spain
2 Institute for Energetics and Interphases-IENI CNR, C.so Stati Uniti 4, 35127 Padova, Italy
Abstract
Optical, electrochemical and spectroelectrochemical features of a family of triisopropylsilyl (TIPS) end-capped oligothienoacenes from the tetramer to the octamer are studied. The oxidation potentials and the formation and stability of the different oxidized species are influenced by the oligothienoacene chain length. The effect of the steric hindrance caused by the substituents on the p-dimerization of the radical cations is also investigated by comparing a trimethylsilyl (TMS) substituted pentathienoacene with a TIPS substituted one.
Keywords: oligothienoacenes, electrochemistry, spectroelectrochemistry.
Introduction
Fully fused a-oligothiophenes, i.e. oligothienoacenes have been drawing extensive attention during the last few years because they combine the molecular shape of linear acenes, molecules widely studied in organic electronics, with thiophene monomer, which may increase stability and nuclear aromaticity and also may provide points of attachment for solubilizing substituents [1,2].
Their rigid fully planar structure without conformational disorder [3-6] generates densely packed structures in solid state (beneficial for reaching high charge-carrier mobility), what makes them ideal candidates to be used in a wide range of electronic and opto-electronic applications, including field effect transistors (FETs) [7-9] and light emitting diodes (LEDs) [10].
However, their structural rigidity leads to reduced solubility with regard to their non-fused a-oligothiophene counterparts. To solve this problem, the most effective synthetic strategies incorporate trimethylsilyl (TMS) and triisopropylsilyl (TIPS) solubilizing substituents at the a-position of thienoacenes [11,12]. On one hand, this end-capping strategy enhances the solubility at the time that lowers the possibility of polymerization under oxidative conditions, which in any case does not occur in our oligothienoacenes because their corresponding radical cations are stabilized by the high degree of p-conjugation. On the other hand, substitution with bulky TMS or TIPS end groups also impedes these oligothienoacenes to achieve close p-p contacts in the solid state, which however constitutes a desiderable feature to assess high charge-carrier mobilities [13-17].
On the basis of different studies, it has been suggested that dimer formation under oxidative conditions is an intrinsic property in unhindered oligothiophenes [18,19]. However, introducing substituents at the b-position of the central ring of oligothiophenes has been employed as a method of steric control of reversible p-dimerization of radical cations [20-22]. To the best of our knowledge, similar studies on a-substituted thienoacenes have not yet been reported in literature.
In the present work a family of triisopropylsilyl oligothienoacenes ranging in length from the tetramer to the octamer (Chart 1) is analyzed by means of optical, electrochemical and spectroelectrochemical techniques, supported by an adequate quantum-chemical study which helps to evaluate the influence of the chain length on the above mentioned properties.
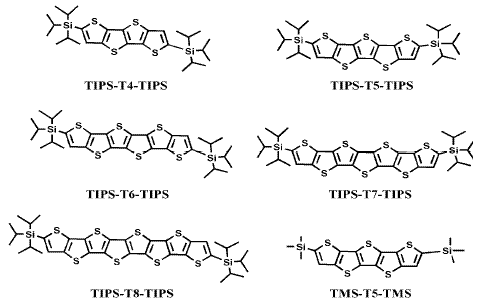
Chart 1
In addition, the effect of the steric interaction of a-substituents on the p-dimer generation upon reversible oxidation is examined through the comparison of two pentathienoacenes bearing different a-substituents.
Experimental
Chemicals and reagents
CH2Cl2 was reagent grade. The supporting electrolyte tetrabutylammonium perchlorate (Bu4NClO4) and all other chemicals were reagent grade and used as received.
A series of triisopropylsilyl a-capped oligothienoacenes with increasing chain length (TIPS-Tn-TIPS, with n = 4-8) and a trimethylsilylpentathienoacene (TMS-T5-TMS) have been prepared according to the literature [23].
Electrochemical apparatus and procedure
Experiments were performed at 25 °C under nitrogen in three electrode cells. The working electrode for cyclic voltammetry was a platinum (0.003 cm2) minidisc electrode; the counter electrode was platinum; the reference electrode was a silver/0.1 M silver perchlorate in acetonitrile. All potentials quoted in the paper are referenced to the ferrocene/ferrocenium couple (Fc/Fc+) as the internal standard. The voltammetric apparatus (AMEL, Italy) included a 551 potentiostat modulated by a 568 programmable function generator and coupled to a 731 digital integrator.
In situ UV-vis-NIR spectroelectrochemistry was performed by controlled-potential electrolysis in a OTTLE cell equipped with a platinum minigrid working electrode and quartz optical windows.
UV–vis absorption spectra were recorded on a Perkin-Elmer Lambda 5 spectrometer.
ESR spectra were taken on a Bruker ER 100D. Temperatures in the range 200-350 K were obtained with a Bruker ER 4111 VT variable temperature unit.
Results and discussion
Optical properties
UV-vis spectra at room temperature of the reported compounds (Fig. 1a) show a partially resolved vibronic structure, a characteristic property of oligothienoacenes, not present in non-fused a-oligothiophenes [24].
s
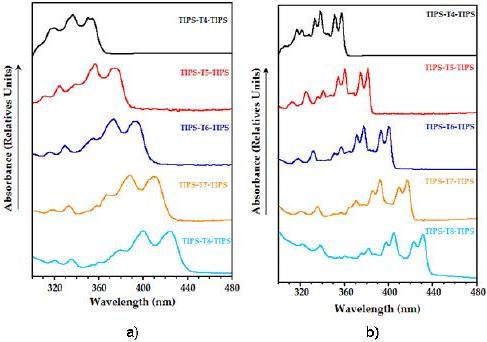
Figure 1. Normalized UV-vis absorption spectra of TIPS-Tn-TIPS (n = 4-8) in MeTHF: a) at room temperature; b) at 93 K.
This feature may be ascribed to the rigid and coplanar molecular structure of heteroacenes, which minimizes the role of the conformational disorder at room temperature. Upon lowering the temperature, the thermal population of torsional modes and the collisions with the surrounding solvent molecules become negligible, thus allowing for electronic absorption spectra to be fully resolved and revealing that only a few vibrational modes couple effectively to the electronic transition (Fig. 1b). All the molecules under study absorb in the near UV region, showing the lowest-energy maximum in the 350-420 nm spectral range (Table 1).
Table 1. Oxidation potentials (E°ox) vs. Fc/Fc+, maximum absorption λexp in THF and TDDFT//B3LYP/6-31G** vertical one-electron excitations related to the strongest UV-vis absorptions of TIPS-Tn-TIPS.
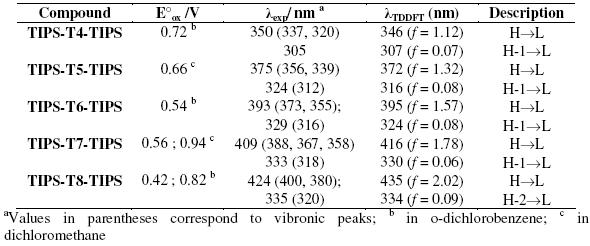
The UV-vis absorption data were computationally interpreted by time-dependent DFT calculations at a B3LYP/6-31G** level. For all the reported systems, these calculations predict the existence of one intense electronic transition in the visible region, which is mainly attributed to a one-electron excitation from the HOMO to the LUMO, together with a second transition at lower wavelength assigned to the one-electron HOMO-1→ LUMO excitation. The calculated electronic transitions are in a very close agreement with experimental data (Table 1).
Electrochemistry and spectroelectrochemistry
All the oligomers exhibit reversible cyclic voltammetric curves (Fig. 2), which demonstrates the presence of a stable cationic species. While the shortest molecules (i.e. tetramer, pentamer and hexamer) present only one oxidation process, the longest ones (heptathienoacene and octathienoacene) display a second wave, indicative of the formation of a dication. Moreover, a third irreversible oxidation process is observed for the heptamer, which indicates the generation of a tricationic species that is largely unstable.
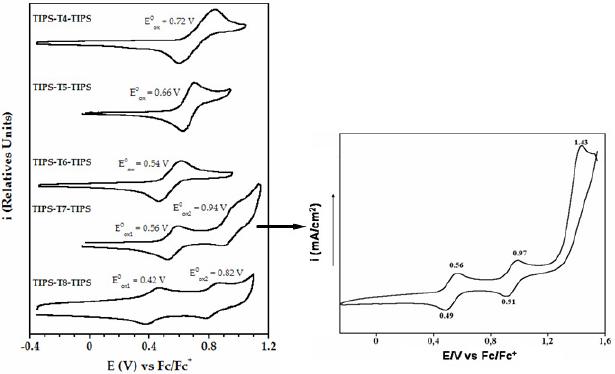
Figure 2. Oxidation potentials and cyclic voltammograms of TIPS-Tn-TIPS (n = 4-8) recorded in a) a solution 0.1 M of nBu4NPF6 in o-dichlorobenzene (n = 4, 6, 8). Scan rate: 0.05 V/s ; b) a solution 0.1 M of Bu4NP in dichloromethane (n = 5, 7). Scan rate: 0.1 V/s.
Spectroelectrochemical analyses confirm the existence of cationic species. Upon the application of an oxidation potential, UV-vis-NIR spectra show the appearance of new absorption bands in the range 500-600 nm and 850-1000 nm (Table 2) typical of radical cations.
Table 2: Maximum absorptions λexp of [TIPS-Tn-TIPS]•+ and [TIPS-Tn-TIPS]++ (n = 4-8).
Further oxidation causes the emergence of new bands at around 700 and 800 nm for the heptamer and octamer respectively, which proves the formation of the dicationic species, stabilized by a more extended p-conjugation.
The oxidation potentials decrease with increasing chain length (Table 1). The longest oligomers are oxidized more easily than the shortest ones because of a more extended p-conjugation.
Substituent effect
Spectroelectrochemical and in-situ ESR analyses
Spectroelectrochemical analyses of the various oligothienoacenes above examined do not reveal the existence of radical cation p-dimers. This fact may be attributed to the presence of the very bulky triisoproylsilyl substituents, responsible for the steric hindrance that avoids for p-dimer formation.
By replacing the TIPS substituents with less voluminous ones (trimethylsilyl, TMS), the steric hindrance is reduced and p-dimer formation is allowed.
With this aim we compared the electrochemical and spectroelectrochemical behaviour of TIPS-T5-TIPS and TMS-T5-TMS.
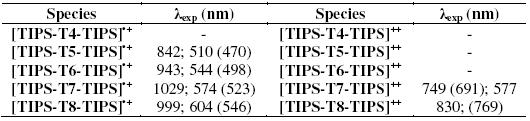
Fig. 3 reports the spectroelectrochemical response for the two compounds. The UV-vis-NIR spectrum of TIPS-T5-TIPS upon oxidation (Fig. 3a) displays two bands at around 500 nm and 800 nm which must be ascribed to the radical cation. TMS-T5-TMS exhibits, after complete electrolysis, (Fig. 3b) one broad band at around 600 nm indicative of the p-dimer generation. Thus, the presence of two bulky TMS end groups, which in principle could be thought to hinder somehow p-dimer formation [25], is not however an obstacle in the present case to the full p-dimerization of the radical cations, a feature which is clearly promoted by the highly planar core of this pentathienoacene.
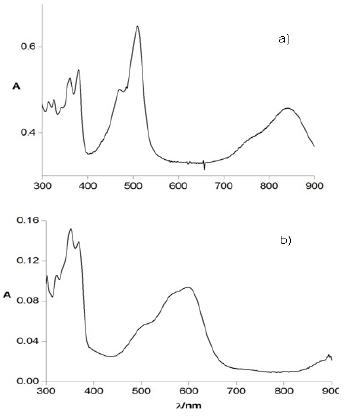
Figure 3. UV-vis spectra in CH2Cl2 + 0.1 M Bu4NClO4 of a) TIPS-T5-TIPS in the radical cation form and b) TMS-T5-TMS in the radical cation dimer form.
The ESR spectrum of a 10-4 M solution of TIPS-T5-TIPS in CH2Cl2 (Fig.4a) after one-electron oxidation displays the signal of the stable radical cation (g = 2.0025). The signal is ca 1 G wide and featureless due to the low number (two) of hydrogen atoms and the relatively high width.
Figure 4. ESR spectra of the radical cations of a) TIPS-T5-TIPS and b) TMS-T5-TMS.
On the contrary, the ESR spectrum of a 10-4 M solution of TMS-T5-TMS in CH2Cl2 after one-electron oxidation (Fig.4b) displays a weak signal which steadily decreases with time, becoming almost undetectable after some hours. This fact agrees with a relatively slow but essentially full p-dimerization.
Conclusions
The whole series of TIPS substituted oligothienoacenes under study exhibit partially resolved electronic absorption spectra at room temperature, which further become fully resolved upon cooling. This spectral feature is due to the lack of molecular disorder, caused by the flat and rigid structure of these fully fused heteroacenes.
Electrochemical properties are also affected by the number of fused heterocycles, as indicated by the experimental oxidation potentials trend, according to which potential values progressively decrease upon increasing chain length. During oxidation of the two longest oligomers (heptamer and octamer), stable dicationic species are generated, while the three shortest oligomers only give rise to the radical cation.
Not only chain length influences the p-dimerization of radical cations upon oxidative doping, but also the nature of the end substituents. By replacing the two bulkier TIPS end moieties with two less bulky TMS ones the steric hindrance between the two interacting pentathienoacene skeletons is significantly lowered, thus allowing for the effective generation of the corresponding radical cation p-dimer.
Acknowledgments
Research at the University of Málaga was supported by the Ministerio de Educación y Ciencia (MEC) of Spain, through project CTQ2006-14987-C02-01, and by the Junta de Andalucía, funding our FQM-0159 scientific group. R.M.O. and C.C.F. are also grateful to MEC for their personal doctoral grants.
References
1. C.D. Dimitrakopoulos, P.R.L. Malenfant, Adv. Mater. 14 (2002) 99. 10.1002/1521-4095(20020116)14:2<99::AID-ADMA99>3.0.CO;2-9
2. G. Horowitz, Adv. Mater. 10 (1998) 365. 10.1002/(SICI)1521-4095(199803)10:5<365::AID-ADMA365>3.0.CO;2-U
3. J. Roncali, Chem. Rev. 97 (1997) 173. 10.1021/cr950257t [ Links ]
4. U. Scherf, J. Mater. Chem. 9 (1999) 1853. 10.1039/a900447e
5. M.D. Watson, A. Fechtenkötter, K. Müllen, Chem. Rev. 101 (2001) 1267. 10.1021/cr990322p
6. M. Bendikov, F. Wudl, D.F. Perepichka, Chem. Rev. 104 (2004) 4891. 10.1021/cr030666m
7. H.E. Katz, Z. Bao, S. L. Gilat, Acc. Chem. Res. 34 (2001) 359. 10.1021/ar990114j
8. C.D. Dimitrakopoulos, P.R.L. Malenfant, Adv. Mater. 14 (2002) 99. 10.1002/1521-4095(20020116)14:2<99::AID-ADMA99>3.0.CO;2-9
9. a) J.E. Anthony, J.S. Brooks, D.A. Eaton, S.R. Parkin, J. Am. Chem. Soc. 123 (2001) 9482; 10.1021/ja0162459
b) M.M. Payne, S.A. Odom, S.R. Parkin, J.E. Anthony, Org. Lett. 6 (2004) 3325; 10.1021/ol048686d
c) M.M. Payne, S.R. Parkin, J.E. Anthony, J. Am. Chem. Soc. 127 (2005) 8028; 10.1021/ja051798v
d) C.R. Swartz, S.R. Parkin, J.E. Bullock, J.E. Anthony, A.C. Mayer, G.G. Malliaras, Org. Lett. 7 (2005) 3163; 10.1021/ol050872b
e) A. Babel, S.A. Jenekhe, J. Am. Chem. Soc. 125 (2003) 13656; 10.1021/ja0371810
f) Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Fukai, Y. Inoue, F. Sato, S. Tokito, J. Am. Chem. Soc. 126 (2004) 8138; 10.1021/ja0476258
g) S. Wakim, J. Bouchard, M. Simard, N. Drolet, Y. Tao, M. Leclerc, Chem. Mater. 16 (2004) 4386; 10.1021/cm049786g
h) Y. Li, Y. Wu, S. Gardner, B.S. Ong, Adv. Mater. 17 (2005) 849. 10.1002/adma.200401290
10. a) J. Jacob, J. Zhang, A.C. Grimsdale, K. Müllen, M. Gaal, E.J.W. List, Macromolecules 36 (2003) 8240-8245; 10.1021/ma034849m
b) J. Jacob, S. Sax, T. Piok, E.J.W. List, A.C. Grimsdale, K. Müllen, J. Am. Chem. Soc. 126 (2004) 6987; 10.1021/ja0398823
c) S. Qiu, P. Lu, X. Liu, F. Shen, L. Liu, Y. Ma, J. Shen, Macromolecules 36 (2003) 9823. 10.1021/ma034929q
11. X. Zhang, A.P. Côté, A.J. Matzger, J. Am. Chem. Soc. 127 (2005) 10502. 10.1021/ja053326m
12. T. Okamoto, K. Kudoh, A. Wakamiya, S. Yamaguchi, Org. Lett. 7 (2005) 5301. 10.1021/ol0523650
13. M.G. Hill, K.R. Mann, L.L. Miller, J.-F. Penneau, J. Am. Chem. Soc. 114 (1992) 2728. 10.1021/ja00033a063
14. a) P. Bäuerle, U. Segelbacher, K.-U. Gaudl, D. Huttenlocher, M. Mehring, Angew. Chem. 105 (1993) 125; Angew. Chem. Int. Ed. Engl. 32 (1993) 76. 10.1002/anie.199300761
15. Y. Yu, E. Gunic, B. Zinger, L.L. Miller, J. Am. Chem. Soc. 118 (1996) 1013. 10.1021/ja9533104
16. G. Brocks, J. Chem. Phys. 112 (2000) 5353. 10.1063/1.481105
17. J.-L. Brédas, J.P. Calbert, D.A. da Silva, J. Cornil, Proc. Natl. Acad. Sci. USA 99 (2002) 5804. 10.1073/pnas.092143399
18. P. Bäuerle, U. Segelbacher, A. Maier, M. Mehring, J. Am. Chem. Soc. 115 (1993) 10217. 10.1021/ja00075a042
19. L.L. Miller, K.R. Mann, Acc. Chem. Res. 29 (1996) 417. 10.1021/ar9600446
20. E. Levillain, J. Roncali, J. Am. Chem. Soc. 121 (1999) 8760. 10.1021/ja990777w
21. J.-M. Raimundo, E. Levillain, N. Gallego-Planas, J. Roncali, Electrochem. Commun. 2 (2000) 211. 10.1016/S1388-2481(00)00007-2
22. D. Yamazaki, T. Nishinaga, N. Tanino, K. Komatsu, J. Am. Chem. Soc. 128 (2006) 14470. 10.1021/ja065995l
23. T. Okamoto, K. Kudoh, A. Wakamiya, S. Yamaguchi, Org. Lett. 7 (2005) 5301. 10.1021/ol0523650
24. G. Macchi, B. Milián Medina, M. Zambianchi, R. Tubino, J. Cornil, G. Barbarella, J. Gierschner, F. Meinardi, Phys. Chem. Chem. Phys. 11 (2009) 984. 10.1039/b810915j
25. P. Bauerle, U. Segelbacherf, A. Maier, M. Mehring, J. Am. Chem. Soc. 115 (1993) 10217. 10.1021/ja00075a042
Received 21 December 2009; accepted 17 March 2010
* Corresponding author: hernandez@uma.es

