










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.2 Porto jun. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i2.17550
WHAT IS YOUR DIAGNOSIS? | QUAL O SEU DIAGNÓSTICO?
Genes, children and pediatricians
Genes, crianças e pediatras
Margarida Paiva CoelhoI, Ana Rita SoaresII, Catarina MagalhãesIII, Ana Teresa VideIV, Esmeralda MartinsI
I. Reference center for Inherited Metabolic Disorders, Centro Materno-Infantil do Norte, Centro Hospitalar Universitário do Porto. 4050-371 Porto, Portugal. margarida.pcoelho@gmail.com; esmeralda.g.martins@gmail.com
II. Centro de Genética Médica, Centro Hospitalar Universitário do Porto. 4099-028 Porto, Portugal. ana.rita.soares@chporto.min-saude.pt
III. Department of Neuropediatrics, Centro Hospitalar Alto Ave. 4810-284 Guimarães, Portugal. catarinadiasmagalhaes@gmail.com
IV. Department of Imaging, Centro Hospitalar Alto Ave. 4810-284 Guimarães, Portugal. ana_teresavide@yahoo.com
Endereço para correspondência | Dirección para correspondencia | Correspondence
Herein is reported the clinical case of a nine-year-old female, second child of a non-consanguineous couple, born at term after an uneventful pregnancy and labor. The girl had regular growth and normal development, except for walking delay acquired at 24 months and recent school complaints about attention issues and reading difficulties. Regarding family history, the father had end-stage kidney disease without known cause and had been transplanted, one female cousin deceased at the age of eight years after central nervous system infection, and another female cousin aged 23 years had a neurodegenerative disease with onset after an infection (Fig. 1).
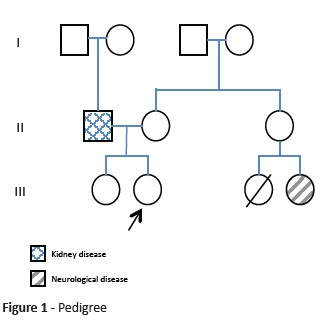
The girl was transferred to the Intensive Care Unit with acute respiratory failure after a 24-hour period of cough, sweating, nocturnal agitation, and ill-being evolving to groan, noisy breathing, and perioral cyanosis. She had experienced respiratory arrest at home, requiring mouth-to-mouth resuscitation. At the emergency room, the girl presented polypnea, hypoxemia, tachycardia, and capillary refill time of four seconds with normal blood pressure, without fever. She was agitated, but neurological exam was otherwise normal. Physical exam showed crackles without wheezing. Leukocyte count was 41.000/ul with low C-reactive protein (9.7 mg/L) and chest x-ray displayed right lung pneumonia signs, for which she started empirical antibiotic treatment. She had progressive respiratory distress with respiratory acidosis and hypotension, requiring mechanical ventilation. Cardiac enzymes, creatine-kinase, and echocardiogram were normal. Microbiological exams were sterile and respiratory secretions were positive for Rhinovirus and Enterovirus.
Clinical evolution was favorable and the patient was extubated at hospitalization day three, after which nystagmus at horizontal gaze extremes, ophthalmoparesis with impaired lateral gaze, ataxia, and discrete dysarthria were noted. Careful symptom review identified episodes of cold extremities, sudoresis and transient skin color alteration, restlessness, irritability, and sleep disturbance in the past month, starting after a sore throat episode.
Brain magnetic resonance revealed symmetrical bilateral T2 hyperintensities on pons and midbrain and basal ganglia involving putamen and caudate (Fig. 2).

(clique para ampliar ! click to enlarge)
What is your diagnosis?
Diagnosis
Leigh syndrome, or subacute necrotizing encephalomyelopathy (MIM # 256000)
Leigh syndrome, or subacute necrotizing encephalomyelopathy (MIM # 256000), is a neurodegenerative disease with characteristic neuroradiological features, including symmetrical T2 basal ganglia hyperintensities with or without brainstem involvement. It translates an energy metabolism disease with more than 75 known causing genes, located either on nuclear or mitochondrial genome.1 It is the most common presentation of mitochondrial disease in childhood, either related with nuclear or mitochondrial genome. Diagnostic criteria include: i) clinical brainstem and/or basal ganglia dysfunction, ii) intellectual and motor developmental delay, and iii) abnormal energy metabolism (severe defect in respiratory chain function or pyruvate dehydrogenase complex activity, molecular diagnosis in a mitochondrial energy-production gene, or elevated serum or CSF lactate).2
Leigh syndrome most often presents after a catabolic event, such as an infection, surgery, or fasting.1 Neurological involvement − as ataxia (basal ganglia) or acute respiratory insufficiency (brainstem involvement) − following such events should raise clinical suspicion of this diagnosis.1-3
In this patient, proband mitochondrial genetic analysis showed mitochondrial ATP synthase subunit 6 gene (MT-ATP6) m.8993T>G mutation in homoplasmy (>95% of mutated mtDNA copies). MT-ATP6 gene codifies for a respiratory chain complex V subunit.3 Further analyses confirmed the same diagnosis in the patient’s cousin.
Acute neurological deterioration with movement disorder or respiratory failure in a patient with a catabolic event (infection with or without fever, surgery, fasting, trauma, etc.) should raise suspicion of Leigh (-like) syndrome.4 In this case, pedigree analysis showing positive family history for individuals interconnected by females raised the possibility of a maternally inherited condition (mitochondrial DNA-related disease). Together with Leigh syndrome diagnosis, a primary mitochondrial disorder related with mitochondrial DNA (mtDNA) was among the first diagnostic options.
Leigh syndrome follow-up includes neurological, ophthalmological (risk of retinopathy), and renal (risk of tubulopathy) evaluation, although other mitochondrial disorder manifestations can occur.4 After six years of follow-up, this patient showed emotional lability, hetero-aggressiveness periods, agitated sleep, and discrete ataxia, with no other brainstem dysfunction or development regression episodes besides developmental delay.
Genetic counseling for mitochondrial-DNA related disorders is extremely challenging. First, phenotype results from a combination of factors, including pathogenic variant severity, percentage of mutated mitochondria (mtDNA heteroplasmy), and tissue distribution, besides potential variants in other relevant genes that can modify phenotype (disease-modifying genes). Consequently, there is a wide phenotypic range even within the same family, ranging from mild to severe symptoms with varying age of onset. This makes virtually impossible to interpret asymptomatic at-risk family member test results in order to have phenotypic prediction. Moreover, absence of mtDNA pathogenic variants in one tissue (e.g. blood) does not guarantee its absence from other tissues (e.g. oocytes).5 Second, risk of recurrence in sibs depends on the mothers’ genetic status, as well as tissue distribution and abnormal mtDNA proportion. Proband mother usually has much lower abnormal mtDNA proportion than the proband and may be asymptomatic.5
Genetic counselling regarding reproductive options is also challenging. Prenatal testing (directly performed on biopsy cells, not cultured) can have conflicting results with an intermediate abnormal mtDNA proportion, with nearly impossible outcome prediction. Oocyte donation accompanied by in vitro fertilization using partner’s sperm can be an option; however, availability of donor oocytes is limited and personal or cultural issues may arise regarding the use of donor gametes. Preimplantation genetic diagnosis is currently considered the preferred reproductive option and can be used to transfer embryos with lower mutant mtDNA load, with low recurrence risk. Mitochondrial donation, in which nuclear material is transferred as pronucleus or maternal spindle from an unfertilized oocyte or single-cell embryo into an enucleated healthy donor cell (with unaffected mitochondria) could potentially block transmission of mutated mtDNA into the developing embryo.5 However, unlike countries such as the United Kingdom, this technique it is not currently available in Portugal.
Overall, genetic counseling for mitochondrial genetic related disease presents considerable challenges. It requires multidisciplinary involvement with an up-to-date experienced team, to ensure that prospective parents are counseled regarding all potential outcomes and limitations of prenatal diagnosis or assisted reproduction.
Keywords: brainstem dysfunction; Leigh syndrome; m.8993T>G; mitochondrial disorder; mtDNA
REFERENCES
1. Ganetzky RD, Stendel C, McCormick EM, Zolkipli-Cunningham Z, Goldstein AC, Klopstock T, et al. Pathogenic variants in MT-ATP6: A United Kingdom-based mitochondrial disease cohort study. Annals of Neurology. 2019; 86:310-5. [ Links ]
2. Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: One disorder, more than 75 monogenic causes. Annals of Neurology. 2016; 79:190-203. [ Links ]
3. Gropman A, Chiaramello A. Phenotypic spectrum of maternally inherited Leigh Syndrome associated with the m.8993T>G variant, Molecular Genetics and Metabolism Reports. 2018; 15:134. [ Links ]
4. Sofou K, de Coo IFM, Ostergaard E, Isohanni P, Naess K, De Meirleir L, et al. Phenotype-genotype correlations in Leigh syndrome: new insights from a multicentre study of 96 patients Journal of Medical Genetics. 2018; 55:21-7. [ Links ]
5. Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. (Assessed September 15, 2019). Available at: https://www.ncbi.nlm.nih.gov/books/NBK1173/. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Margarida Paiva Coelho
Reference center for Inherited Metabolic Disorders
Centro Materno-Infantil do Norte
Centro Hospitalar Universitário do Porto
Rua da Maternidade
4050-371 Porto, Portugal.
Email: margarida.pcoelho@gmail.com
Received for publication: 25.03.2019. Accepted in revised form: 16.03.2020

