










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.2 Porto jun. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i2.15028
CASE REPORTS | CASOS CLÍNICOS
Behçet’s syndrome in pediatric age
Síndrome de Behçet em idade pediátrica
Ana Raquel MendesI, Sandrina BragaII, Catarina VilarinhoIII, Maria Antónia CostaIV, Cristina FerreiraV, Teresa São SimãoV
I. Department of Pediatrics, Centro Materno-Infantil do Norte, Centro Hospitalar Universitário do Porto. 4050-651 Porto, Portugal. ana.mendes1890@gmail.com
II. Department of Vascular Surgery, Hospital da Senhora da Oliveira. 4835-044 Guimarães, Portugal. sandrina.figueiredo.braga@gmail.com
III. Department of Dermatology, Hospital da Senhora da Oliveira. 4835-044 Guimarães, Portugal. catvilarinho@gmail.com
IV. Department of Ophtalmology, Hospital da Senhora da Oliveira. 4835-044 Guimarães, Portugal. mariantonia.oft@gmail.com
V. Department of Pediatric, Hospital da Senhora da Oliveira. 4835-044 Guimarães, Portugal. crispediatra@gmail.com; teresinhaped@gmail.com
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
Introduction: Behçet´s syndrome is a systemic vasculitis characterized by recurrent oral and/or genital ulcers, and several systemic manifestations. The authors describe the case of a pediatric-onset Behçet´s syndrome.
Case report: An 11-year-old boy was referred to the Pediatric consultation after two episodes of great saphenous vein thrombophlebitis. He had experienced daily oral aphthae for the past three years, and various episodes of folliculitis with pustule formation. Laboratory study was normal. The boy showed no signs of uveitis. The diagnosis of Behçet´s syndrome diagnosis was established according to the international criteria, with positive HLAB51 testing. Colchicine was initiated, with favourable response.
Conclusions: Due to clinical feature overlap with other conditions, Behçet’s syndrome diagnosis remains challenging. Consensus pediatric classification criteria developed in 2016 enabled greater sensitivity and earlier diagnosis.
Keywords: Behçet´s syndrome; classification criteria; pediatric age
RESUMO
Introdução: A síndrome de Behçet é uma vasculite caracterizada por episódios recorrentes de aftas orais e/ou genitais e manifestações sistémicas diversas. Os autores descrevem um caso de Síndrome de Behçet em idade pediátrica.
Caso clínico: Um adolescente de 11 anos foi referenciado à consulta de Pediatria após dois episódios de tromboflebite da veia safena magna. Reportou episódios recorrentes de lesões aftosas nos últimos três anos e vários episódios de foliculite com a formação de pústulas. O estudo analítico foi normal. O exame oftalmológico não demonstrou sinais de uveíte. Foi diagnosticada Síndrome de Behçet de acordo com critérios internacionais. O estudo genético foi positivo para o antigénio HLA-B51. Foi iniciada colchicina, com resposta favorável.
Conclusões: Devido à sobreposição de características clínicas com outras condições, o diagnóstico de Síndrome de Behçet permanece um desafio. Os critérios de classificação em idade pediátrica, elaborados em 2016, permitiram uma maior sensibilidade e diagnóstico mais precoce.
Palavras-chave: critérios de classificação; idade pediátrica; síndrome de Behçet
Introduction
Behçet´s syndrome (BS), also known as Behçet´s disease, is a systemic vasculitis involving blood vessels of any diameter. It is characterized by recurrent oral ulcers affecting the oral and genital mucosa and skin lesions. It also comprises several systemic manifestations, including ocular, neurological, cardiovascular or gastrointestinal involvement, and arthritis. Among systemic vasculitides, BS stands out for its ability to affect both venous and arterial blood vessels of all sizes.1,2
Although previously defined as a very rare entity, recent studies show that BS is more frequent than initially suspected. Its prevalence ranges from 5.2 to 7.1 cases per 100,000 adults in the USA and France, respectively.3,4 Overall prevalence is similar between men and women. The disease typically affects young adults aged 20 to 40 years, but in 4−26% of cases it is also observed in children before the age of 16.5
Although BS has been classified as a vasculitis, its underlying cause remains unclear. Aberrant immune activity triggered by a specific agent in patients with genetic predisposition has been suggested as the underlying pathogenesis.6 A number of disease mechanisms, including genetic predisposition (e.g. association with certain human leukocyte antigens (HLA) and non-HLA genes), altered host bacteria response, altered hematopoietic stem cell populations and respective cytokines, presence of immune complexes and autoantibodies, and vascular endothelial activation and hypercoagulability have been suggested. Triggers may include viral or bacterial antigens or other environmental materials, such as chemicals or heavy metals.6
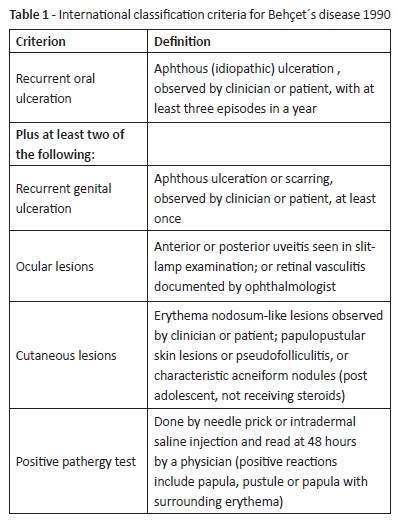
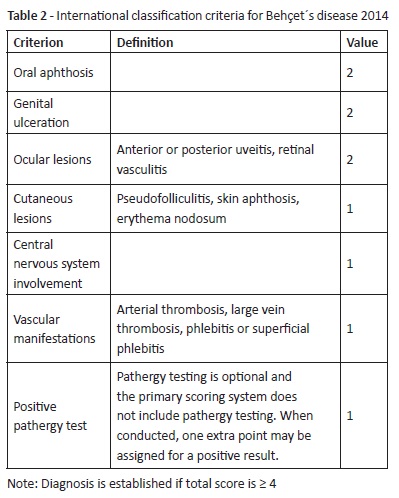
BS diagnosis is based on clinical features. The most used classification was proposed by the International Study Group for Behçet´s disease in 1990 and includes recurrent oral ulceration at least three times a year as mandatory feature, along with at least two additional minor criteria (Table 1).7 Low criteria sensitivity led to a revision in 2014, which added neurological and vascular features to criteria (Table 2).8 Still, a validated, sensitive, and specific BS definition in pediatric age remains absent.9
The authors present a case of pediatric-onset Behçet´s syndrome with HLA-B51 positivity.
Case report
An 11-year-old boy with a history of recurrent oral aphthae was admitted to the Emergency Department due to pain, redness, and warmth on the inner surface of the left leg with two weeks of evolution, without fever. On physical examination, painful swelling along a venous path on the left leg with inflammatory signs was identified. Dorsalis pedis pulse was bilaterally palpable. A lower limb echo-Doppler ultrasound confirmed thrombophlebitis of the great superficial saphenous vein. The boy was discharged with enoxaparin 20 mg for two weeks and referred to outpatient consultation. After almost four months with no intercurrences, he was readmitted to the Emergency Department due to a second episode of swelling, redness, and warmth on the inner surface of the left leg. On physical examination, the patient showed local inflammatory signs without gait impairment and several oral aphthae. Case was discussed with a vascular surgeon and the patient was medicated with another course of enoxaparin 20 mg for two weeks and discharged with recommendation of rest, lower limb elevation, and oral hydration.
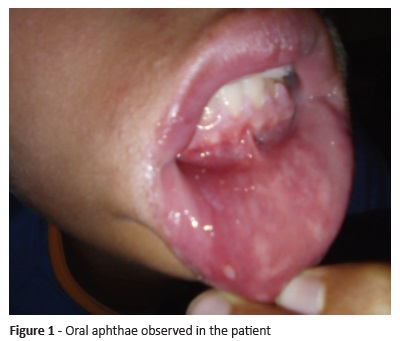
In the first Pediatric outpatient consultation, the patient reported a history of daily painful oral aphthae (Figures 1 and 2) with spontaneous resolution after ten days for the past three years (more than three episodes a year). His mother described several episodes of lower limb folliculitis with pustule formation over the previous months. She also referred episodes of erythema and pain in the metatarsophalangeal joints of the right toe.
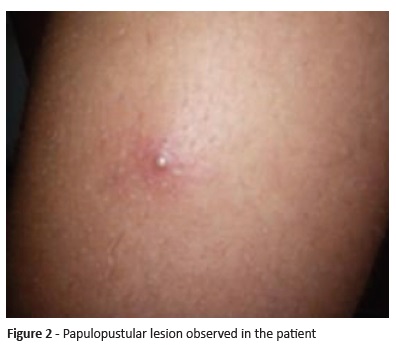
The patient denied fever, rash, weight gain or loss, syncope, sleep disturbance, headache, abdominal pain, change in bowel habits or bladder function, respiratory symptoms, visual disturbances, or fatigue. Recent infections were not reported. Physical examination was normal, except for two oral aphthae and some acneiform lesions, prompting more accurate investigation of systemic lupus erythematosus and Behçet ´s syndrome.
Laboratory study included complete blood count with renal and hepatic function, C-reactive protein, coagulation study, hepatitis B and C and HIV viral markers, herpes simplex and varicella-zoster virus serology, and calprotectin levels, which were all negative. Immunoglobulin levels were normal and autoimmunity study including rheumatoid factor, antinuclear antibodies, anti-cardiolipin and beta 2 glycoprotein antibodies, lupus anticoagulant, anti-extractable nuclear antigen (ENA), anti-neutrophil cytoplasmic (ANCA) antibodies, and celiac disease markers were negative. Urine analysis revealed discrete proteinuria and pathergy test was negative.
On Dermatology consultation five months after the initial vascular event, no findings were documented except for some acneiform lesions. Echocardiogram was normal and ophthalmologic evaluation showed no signs of uveitis.
According to the International Criteria for Behçet´s disease 2014 (Table 2), the patient scored four (recurrent oral aphthosis, cutaneous lesions, and vascular signs) and was diagnosed with Behçet´s syndrome. HLAB51 test, which was later requested, was positive.
Approximately one year after the first vascular event, the patient reported a genital ulcer episode, adding another item to the international clinical diagnostic criteria.
After diagnosis, he was prescribed oral colchicine 1.5 mg per day, with subsequent reduction of oral aphthous lesion number and size. No further occurrences were reported, except for one self-limited epididymitis episode.
Discussion
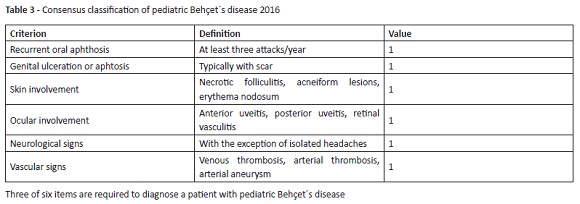
Due to clinical feature overlap with several other conditions, especially autoinflammatory diseases, inflammatory bowel diseases, and immunodeficiencies, BS diagnosis remains challenging.9 As oral aphthous ulcers are common in the general population compared with BS, which has a relatively low incidence, diagnosis should be suspected in presence of recurrent aphthous ulcers along with characteristic systemic manifestations, particularly ocular lesions and neurological and vascular disease. No pathognomonic laboratory for the condition is in place and diagnosis is based on clinical findings. Consensus classification criteria have been recently developed for pediatric Behçet´s syndrome (Table 3), in an attempt to increase prior criteria sensitivity.9
Mucocutaneous lesions remain the main clue for BS diagnosis. Ocular lesions are the second most prominent lesion type, occurring in 25-75% of patients.1 Bilateral episodic uveitis is the dominant feature.10 As in the present case, approximately 20−40% of children report articular involvement, generally arthralgia, mostly in the knees, ankles, elbows, and wrists. Neurological manifestations include acute features, such as recurrent aseptic meningitis and meningoencephalitis, and chronic features, such as progressive parenchymal and vascular lesions. The latter usually manifest as cognitive impairment, memory loss, depression, anxiety, pseudobulbar syndrome, and sensitive and motor deficits. The most common presentations in pediatric age are cerebral venous thrombosis and cranial nerve palsy, especially of the VIth nerve.1
Most BS clinical manifestations are attributed to vasculitis, which can affect both arterial and venous blood vessels of all sizes. Prevalence of pediatric vascular disease was reported to range from 9.6% to 15% in two case series and have a high relapse rate, as observed in this case.5,11
Although ileum and colon are the most commonly affected intestinal sites, all gastrointestinal tract can be affected. Isolated abdominal pain, diarrhea, and bleeding are among the most prevalent symptoms.1
In BS, renal involvement is less severe and less frequent compared with other vasculitides. Patients may present proteinuria, as in the present case, hematuria, or mild renal insufficiency. The first two may occur in up to 10% of patients.12
Cardiac manifestations include pericarditis, myocarditis, and intracardiac thrombus. Orchitis, epididymitis, urethritis, and sterile cystitis cases, although rare, have been described.1
A study conducted by Koné-Paut et al. in 1999 reported a significantly higher frequency of familial cases in pediatric compared with non-pediatric BS patients, suggesting a strong genetic component.13 Presence of the HLA-B51 antigen, as in this case report, has been associated with an increased risk of BS and disease severity. Nevertheless, absence of HLA-B51 antigen does not exclude BS.1
BS has a recurrent and unpredictable course, often remaining active in children with new symptom onset over time. Morbidity is usually related to neurological and ocular disease.1 Mortality, on the other hand, is associated with vascular disease, malignancy, central nervous system involvement, and sepsis. Younger age, male gender, high flare number, and arterial involvement are risk factors for death.14 Consequently, these patients require periodic ophthalmological evaluation and close monitoring of prothrombotic states.
No specific treatment is established for BS. Treatment goal is suppression of inflammatory exacerbations and recurrences to prevent irreversible organ damage. Following the 2018 update of the European League Against Rheumatism (EULAR) recommendations for BS management, the present patient was prescribed colchicine for preventing recurrent mucocutaneous lesions, with clinical improvement. Use of topical steroids is recommended for oral and genital ulcer treatment, as well as for papulopustular or acne-like lesions. Glucocorticoids and immunosuppressive therapy, such as azathioprine, cyclophosphamide, or cyclosporine A, are recommended for acute deep vein thrombosis management.15 Since this patient only had superficial venous thrombotic events, none of these agents was employed.
REFERENCES
1. Koné-Paut I. Behçet´s disease in children, an overview. Pediatric Rheumatology. 2016; 14:10. [ Links ]
2. Kari JA, Shah V, Dillon MJ. Behçet´s disease in UK children: clinical features and treatment including thalomide. Rheumatology 2001; 40:933-8. [ Links ]
3. Mahr A, Belarbi L, Wechsler B, Jeanneret D, Dhote R, Fain O, et al. Population-based prevalence study of Behçet´s disease: differences by ethnic origin and low variation by age at immigration. Arthritis Rheum 2008; 58:3951-9. [ Links ]
4. Calamia KT, Wilson FC, Icen M, Crowson CS, Gabriel SE, Kremers HM. Epidemiology and Clinical Characteristics of Behçet´s disease in the US: a population-based study. Arthritis Rheum 2009; 61:600-4. [ Links ]
5. Karincaoglu Y, Borlu M, Toker SC, Akman A, Onder M, Gunasti S, et al. Demographic and clinical properties of juvenile-onset Behçet´s disease: A controlled multi-center study. J Am Acad Dermatol 2008; 58:579-84. [ Links ]
6. Direskeneli H. Behçet´s disease: infectious aetiology, new autoantigens, and HLA-B51. Ann Rheum Dis 2001; 60:996-1002. [ Links ]
7. International Study group for Behçet´s disease. Criteria for diagnosis of Behçet´s disease. Lancet. 1990; 335:1078-80. [ Links ]
8. International Team for the Revision of the International Criteria for Behçet´s disease (ITR-ICBD). The International Criteria for Behçet´s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014; 28:338-47. [ Links ]
9. Koné-Paut I, Shahram F, Darce-Bello M, Cantarini L, Cimaz R, Gattorno M, et al. Consensus classification criteria for paediatric Behçet´s disease from a prospective observational cohort: PEDBD. Ann Rheum Dis. 2016; 75:958-64. [ Links ]
10. Kokturk A. Clinical and Pathological Manifestations with Differential Diagnosis in Behçet´s disease. Patholog Res Int. 2012; 2012:690390. [ Links ]
11. Koné-Paut I, Yurdakul S, Bahabri SA, Shafae N, Ozen S, Ozdogan H, et al. Clinical features of Behçet´s disease in children: an international collaborative study of 86 cases. J Pediatr. 1998; 132:721-5. [ Links ]
12. Altiparmak MR, Tanverdi M, Pamuk ÚN, Tunç R, Hamuryudan V. Glomerulonephritis in Behçet´s disease: Report of seven cases and review of literature. Clin Rheumatol. 2002; 21:14-8. [ Links ]
13. Koné-Paut I, Geisler I, Wechsler B, Ozen S, Ozdogan H, Rozenbaum M, et al. Familial aggregation in Behçet´s disease: high frequency in siblings and parents of pediatric probands. J Pediatr. 1999; 135:89-93. [ Links ]
14. Saadoun D, Wechsler B, Desseaux K, Le Thi Huong D, Amoura Z, Resche-Rigon M, et al. Mortality in Behçet´s disease. Arthritis Rheum. 2010; 62:2806-12. [ Links ]
15. Hatemi G, Christensen R, Bang Dongsik, Bodaghi B, Celik AF, Fortune F, et al. 2018 Update of the EULAR recommendations for the management of Behçet´s syndrome. Ann Rheum Dis 2018; 77:808-18.
Endereço para correspondência | Dirección para correspondencia | Correspondence
Ana Raquel Mendes
Department of Pediatrics
Centro Materno-Infantil do Norte
Centro Hospitalar Universitário do Porto
Largo da Maternidade
4050-371 Porto, Portugal.
Email: ana.mendes1890@gmail.com
Received for publication: 10.09.2018. Accepted in revised form: 02.10.2019

