










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.27 no.4 Porto dez. 2018
https://doi.org/10.25753/BirthGrowthMJ.v27.i4.13021
CASE REPORTS | CASOS CLÍNICOS
Linear IgA bullous dermatosis: report of an exuberant clinical case and literature review
Dermatose bolhosa iga linear: relato de um caso exuberante e revisão da literatura
Sandra PereiraI, Alexandra MartinsII, Teresa OliveiraII, Virgínia MonteiroII
I Pediatrics Department, Hospital Pediátrico Integrado. Centro Hospitalar Universitário São João. 4200-319 Porto, Portugal. sandravdpereira@gmail.com
II Pediatrics Department, Centro Hospitalar Entre Douro e Vouga. 4520-211 Santa Maria da Feira, Portugal. lmartins.alexandra@gmail.com; teresapoliveira@gmail.com; virginiacostamonteiro@gmail.com
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
Introduction: Linear IgA dermatosis (LIGAD) is a rare acquired disease, with a probable autoimmune origin. Its differential diagnosis involves other bullous dermatosis.
Case Report: A previously healthy 12-month-old male was observed at the Emergency Department due to a 4-day itchy vesiculobullous rash in the perineal region, lower abdomen, hands, and feet. Analytical study was normal. Flucloxacillin and hydroxyzine were initiated without improvement. New (some of which confluent) lesions, erosions, and serohematic crusts developed on the periphery of previous lesions. A skin biopsy was performed at this time, revealing a subepidermal blister with neutrophilic infiltrate at histological examination. Direct immunofluorescence uncovered linear IgA deposits along the basement membrane. Lesion remission occurred without further therapeutic measures.
Discussion: Although clinically exuberant, LIGAD is usually a self-limited disease. A high degree of suspicion is important, since immunofluorescence is diagnostic and pathognomonic, avoiding late diagnosis, unnecessary treatments, and parental anxiety.
Keywords: Linear IgA bullous dermatosis; pediatrics; skin diseases; vesiculobullous
RESUMO
Introdução: A dermatose IgA linear (DIGAL) infantil é uma doença adquirida rara, de origem autoimune provável, que implica diagnóstico diferencial com outras dermatoses bolhosas.
Caso Clínico: Uma criança de 12 meses, saudável, do género masculino, foi observada no Serviço de Urgência por erupção vesico-bolhosa, pruriginosa, com quatro dias de evolução, na região perineal, abdominal inferior, mãos e pés. O estudo analítico não revelou alterações. A criança iniciou flucloxacilina e hidroxizina, sem melhoria. Verificou-se o aparecimento de novas lesões (algumas confluentes) na periferia das antigas, assim como erosões e crostas serohemorrágicas. Foi efetuada uma biópsia cutânea, cujo exame histológico revelou bolha subepidérmica com infiltrado neutrofílico. A imunofluorescência direta revelou depósitos lineares de IgA ao longo da membrana basal. Observou-se evolução com remissão das lesões sem medidas terapêuticas adicionais.
Conclusão: A DIGAL, apesar de clinicamente exuberante, é geralmente autolimitada. É necessário um elevado índice de suspeição, já que a imunofluorescência é diagnóstica e patognomónica, permitindo evitar diagnósticos tardios, tratamentos desnecessários e ansiedade parental.
Palavras-chave: Dermatose bolhosa IgA linear; pediatria; doenças cutâneas; vesico-bolhoso
Introduction
Autoimmune vesiculobullous diseases of the skin are rare in children, with linear immunoglobulin (Ig) A dermatosis (LIGAD) being the most common autoimmune bullous dermatosis at this stage of life.1-4 Pediatric prevalence of LIGAD is unknown.5 It is an acquired bullous disease, probably of autoimmune etiology, which manifests by the presence of vesiculobullous eruption, erosions and/or secondary crusts, usually limited to the skin but possibly also involving the mucosa.1,2,4,6-9 It can be clinically misdiagnosed as dermatitis herpetiformis, bullous pemphigoid, bullous impetigo, or epidermolysis bullous. Therefore, when a child presents with bullous dermatosis, a diagnosis of LIGAD should be considered.1,2,6,7 Histologically, it is characterized by presence of a subepidermal cleavage with neutrophilic and eosinophilic inflammatory infiltrate.1,2,5,7,10
The advent of direct immunofluorescence (DIF) in the 1970’s allowed for a precise diagnosis of the disease by identifying linear IgA antibody deposition in the basement membrane, which is pathognomonic of LIGAD.1,2,5-7,9-11 Secondary dermatosis results from development of IgA antibodies against normal basement membrane components, although the target antigen is not fully identified yet.1 Antibodies are found either in the skin or blood.1,2
Case report
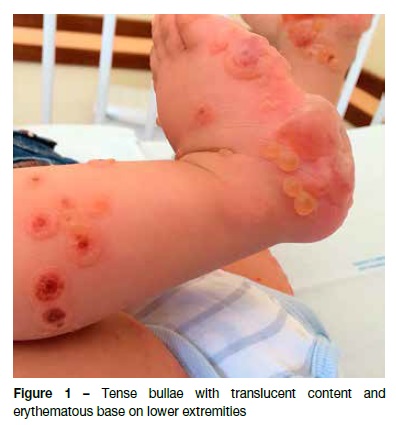
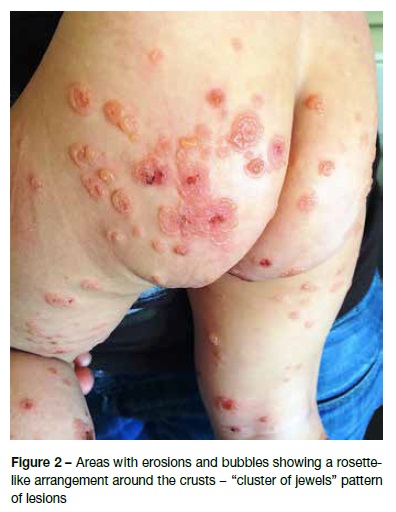
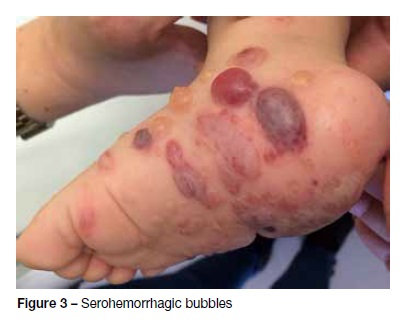
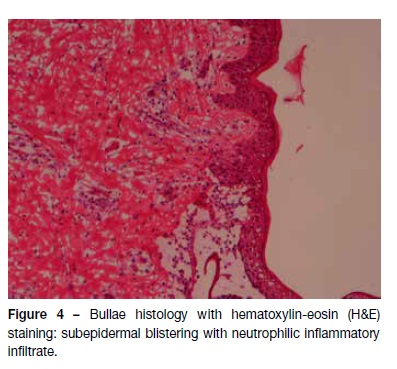
A 12-month-old male, with irrelevant personal or family background, was observed in the Emergency Department due to vesiculobullous eruption with intense pruritus developing four days earlier. It was initially detected in the perineum and progressed to the trunk and limbs. The infant had no history of infections, trauma, or recent medications, or any other associated symptoms. Physical examination disclosed vesicles and tense bullous of citrus content (Figure 1), with negative Nikolsky’s sign, some of which on normal skin and others on an erythematous base. They were symmetrically distributed and located in the perineum, lower abdominal quadrants, and extremities, some of which with local inflammatory signs. Areas of erythematous skin surrounded by bullous were also visible, in a pattern of “cluster of jewels”, with a rosette-like arrangement around the crusts (Figure 2). Blood tests, including complete blood count, hepatic and renal function were normal and rapid antigen Group A Streptococcus test was negative. Due to suspicion of infectious pathology (possibly bullous impetigo), flucloxacillin and hydroxyzine were initiated, and the child was admitted to the Pediatric Department. No improvement was observed after 48 hours, but instead new lesions were observed in the periphery of old ones − some of which confluent and others grouped in a “cluster of jewels” −, as well as erosions and serohemorrhagic blisters (Figure 3). No perioral or ocular attainment was observed. Autoimmune dermatosis skin biopsies were also performed. Histological examination showed a subepidermal blister with neutrophilic infiltrate in the dermal papillae (Figure 4) and direct immunofluorescence confirmed the diagnosis of LIGAD by showing a linear arrangement of IgA along the basement membrane. As from the sixth day of hospitalization, and without institution of other therapeutic measures, a significant lesion improvement was observed. The infant completed 14 days of antibiotic therapy and was discharged upon referral to the Dermatology and Pediatrics consultations, remaining in complete remission after 12 months of follow-up.
Discussion
LIGAD is a bullous dermatosis with probable autoimmune etiology, resulting from IgA antibodies elicited against basement membrane components.1,2,4,10,12,13 It has a worldwide distribution, being more prevalent in developing countries and in the female gender.2,3,6,9 Although target antigen is not fully identified, IgA antibodies are deposited on the basement membrane of all patients at the macroscopic lesion site, and circulating antibodies are detected in most cases.1,2 Through western immunoblot is possible to characterize potential antigens, particularly of 97 kDa (LABD-97) and 120 kDa (LAD-1), which are located in the lucid lamina and represent fragments of the extracellular domain of collagen XVII (BP180), bullous pemphigoid antigen 2 (BPAG2, 180 KDa).2,3,6,8,11,14 Other antigens, such as collagen VII (250 kDa) and BP230 (BPAG1, 230 kDa) have also been recognized.3,8,14 Although no precipitating factor has been identified in this patient, LIGAD may be triggered by such diverse causes as prodromal diseases − including upper airway (Epstein-Barr virus and cytomegalovirus) or urinary tract infections −, brucellosis, acute gastroenteritis (Salmonella enteritis), tetanus, or immunizations (varicella or influenza vaccine).1,2,15 Drug-induced LIGAD, although less frequent in pediatric ages, is more atypical and severe than spontaneous forms, with lesions mimicking toxic epidermal necrolysis and positive Nikolsky’s signal.11 Cases associated with antibiotics (most frequently vancomycin, but also ceftriaxone, amoxicillin-clavulanic acid, and trimethoprim-sulfamethazol), amiodarone, non-steroidal anti-inflammatory agents, and diuretics such as captopril have also been reported.1-3,5,6,9,11,12,14
In the case of a three-year-old boy, identification of trimethoprim-sulfamethazol as the causative drug was accomplished after rechallenge. Lesion resolution occurred spontaneously within two to seven weeks after medication withdrawal.5 Most studies do not confirm a correlation with autoimmune diseases, but associations with Celiac Disease, Crohn’s Disease, Ulcerative Colitis, and Sjogren’s Syndrome have been reported1,5,7, as well as with lymphoproliferative diseases and solid tumors (carcinoma of bladder).7
LIGAD frequently presents with cutaneous-mucosal attainment, without affecting other organs or systems.1,4 It usually has an abrupt onset and occurs in children after the age of six months, with a peak incidence at pre-school age (four to five years). Development of vesicles and stray blisters on the apparently normal or erythematous base of the skin is observed after non-specific pruritus or burning prodromes.1-6,9,14,15 However, cases of LIGAD in newborns, with involvement of (particularly ocular) mucous membranes and more severe clinical presentations have been reported.4,15 Systemic symptoms, such as fever and anorexia, may occur and have an insidious onset.1 Development of new tense vesicles at the periphery of old lesions is common, usually grouping in rosette or “cluster of jewels”.1-4,8,9,11,13,14 It typically affects the perineum and lower abdomen, extending to the thorax and extensor surface of the limbs1-4, as in the reported clinical case.
Involvement of mucous membranes manifests by conjunctivitis and oral and nasal erosions.4,13 Perioral lesions are difficult to distinguish from aphthous ulcers, although they appears less frequently than in the adult form.1-3,6 An ocular examination should be performed, due to risk of subconjunctival fibrosis, symblepharon, trichiasis, cicatricial entropion and, rarely, corneal opacities.2-4, 8
In the present clinical case, the child did not present ocular symptoms and ocular examination was normal. It should be noted that there is no correlation between severity and disease duration.1 Because this is clinically indistinguishable from other autoimmune bullous dermatoses, the differential diagnosis should include bullous pemphigoid and dermatitis herpetiformis as main diagnoses to be considered, as well as acquired epidermolysis bullosa and bullous lupus erythematosus.1,2,7-9,13,15 When lesions are limited to the perineal region, bullous impetigo and herpes simplex should be considered.1
Dermatitis herpetiformis is characteristically associated with sensitive enteropathy to gluten and presents with complaints of more intense pruritus, more diffuse neutrophilic infiltration, granular deposition of IgA on the basement membrane, and detection of anti-gliadin and anti-transglutaminase antibodies.2 On the other hand, bullous pemphigoid usually occurs in older age groups, shows a predominantly eosinophilic inflammatory infiltrate, linear deposition of IgG and C3 in the basement membrane at DIF, and detection of anti-BP180 and anti-BP230 antibodies.2 As LIGAD is a rare disease, usually late diagnosed, high suspicion is necessary, since an early biopsy is confirmatory.1,2
Delayed diagnosis predisposes to lesion secondary infection and, less frequently, to comorbidities such as ocular scarring and pharyngolaryngeal stenosis.3 Histologically, subepidermal blisters are observed in association with neutrophilic and, sometimes, eosinophilic infiltrate, which is also observed in bullous pemphigoid and dermatitis herpetiformis.1-3,6,9,13 A definitive diagnosis is possible through DIF, which is pathognomonic and evidences the linear deposits of IgA on the basement membrane.1,2,13 Therapy is symptomatic, and when triggering factors are identified, their disruption is key.1,2 In most exuberant cases, first-line treatment for bullous lesions consists of dapsone, with sulfonamides also being an option.1-3,9,10,13,15
Dapsone acts by interfering with folate biosynthesis and neutrophil activity and is used at an initial dose of 1 to 2 mg/Kg/day. Response occurs rapidly (within 24 to 48 hours) and, depending on the patient’s tolerability, a gradual dose increase is recommended.1,3 The drug’s use is limited by side effects, such as hemolysis, methemoglobinemia, neutropenia, motor neuropathy, and hepatitis, reason why a blood count should be performed weekly during the first month, monthly during the 2nd to 5th months, and then twice a year after the first six months. Renal and hepatic function should also be controlled at the 6th month of treatment and then annually.1,2,13 Glucose-6-phosphate dehydrogenase deficiency should be excluded before starting treatment, due to risk of severe hemolytic anemia,1-3,9,13,15 as well as severe hemolytic anemias, bone marrow suppression, and dapsone hypersensitivity syndrome.2,15 Duration of treatment should be individualized, although most clinicians keep patients on maintenance treatment for 3−21 months.8 Sulphapyridine at 35 mg/kg/day may be alternatively used and blood count monitoring should be performed.2
Prednisolone is administered at 0.5−2 mg/kg/day and dose should be gradually reduced two weeks after lesions resolved. It is not usually used as a first-line option, but as an adjunct to dapsone or sulphapyridine.1,2,8,9,13 However, cases with good clinical response to corticosteroids alone, especially in drug-induced forms, have been described.2 Alternative therapy with colchicine is indicated only in exceptional cases, for patients who are not candidates for sulfones.1,2 Mycophenolate mofetil may be used as monotherapy or in combination, as a corticosteroid sparing agent.1,2
Sporadic success cases with antibiotics have been reported, including with flucloxacillin, dicloxacillin, erythromycin, and cotrimoxazole.2,3,8,9,15 As flucloxacillin was initiated in the present clinical case, it is not possible to determine whether resolution was spontaneous or influenced by this antibiotic therapy. Cases have been reported in the literature of rapid and long-term remissions with flucloxacillin, but also of the drug’s ineffectiveness.2,15 Its use requires hepatic monitoring due to risk of cholestatic hepatitis.2 Intravenous immunoglobulin should be considered for patients refractory to these treatments, at a dose of 1−2 g/kg monthly in 3-day cycles.2,13 Studies describing the use of intravenous immunoglobulin reported average treatment durations ranging from months to years.7,8 Although LIGAD is a benign condition, it often presents with recurrences until complete resolution. It may also be self-limited, with remission and no sequelae in two to four years.2,3,9,10,15 In absence of infections or itching, cutaneous lesions heal without scarring.8,9 Persistence after puberty is rare, particularly in adulthood.1,3 Most cases require pharmacological treatment and its early initiation (within the first month) may be a decisive factor in inducing an early remission.2,8,15
This case illustrates an exuberant presentation of an uncommon childhood illness, with a favorable response without dapsone or corticosteroid therapy. The authors intend to raise awareness to this entity and stress the importance of an early diagnosis and adequate parental guidance to avoid untimely treatment, distress, and parental anxiety.
REFERENCES
1. Rocha F, Silva A, Fonseca P, Teixeira P, Vieira AP, Oliveira JMG. Dermatose Bolhosa Crónica Linear por IgA: Apresentação de um Caso Clínico e Revisão da Literatura. Acta Pediatr Port. 2004; 35:521-4. [ Links ]
2. Ferreira O, Mota A, Morais P, Duarte AF, Bettencourt H, et al. Dermatose IgA linear da Infância - Apresentação em cacho de uva escrotal. Revista SPDV 2011; 69:621-5. [ Links ]
3. Souza BC, Fregonesi NC, Tebcherani AJ, Sanchez AP, Aoki V, et al. Linear IgA bullous dermatosis: report of an exuberant case. An Bras Dermatol 2013. 88:67-70. [ Links ]
4. Magalhães JC, Oliveira AC, Machado S, Reis MG. Dermatose bolhosa IgA linear. Acta Pediatr Port. 2013; 44:46-7. [ Links ]
5. Alajlan A, Al-Khawajah M, Al-Sheikh O, Al-Saif F, Al-Rasheed S, Al-Hoqail I, et al. Treatment of linear IgA bullous dermatosis of childhood with flucloxacillin. J Am Acad Dermatol. 2006; 54:652-6. [ Links ]
6. Romani L, Diociaiuti A, D’Argenio P, El Hachem M, Gargiullo L, Boldrini R, et al. A Case of Neonatal Linear IgA Bullous Dermatosis with Severe Eye Involvement. Acta Derm Venereol. 2015 4; 95:1015-7.
7. Lings K, Bygum A. Linear IgA bullous dermatosis: a retrospective study of 23 patients in Denmark. Acta Derm Venereol. 2015; 95:466-71. [ Links ]
8. Reyes-Baraona F, Andino R, Carrasco JE, Arriagada C, Guerrero S. Linear IgA bullous dermatosis of childhood: case report. Arch Argent Pediatr. 2014; 112:e57-60. [ Links ]
9. Chanal J, Ingen-Housz-Oro S, Ortonne N, Duong TA, Thomas M, Valeyrie-Allanore L, et al. Linear IgA bullous dermatosis: comparison between the drug-induced and spontaneous forms. Br J Dermatol. 2013; 169:1041-8. [ Links ]
10. Venning VA. Linear IgA disease: clinical presentation, diagnosis, and pathogenesis. Immunol Allergy Clin North Am. 2012; 32:245-53. [ Links ]
11. Kharfi M, Khaled A, Karaa A, Zaraa I, Fazaa B, Kamoun MR. Linear IgA bullous dermatosis: the more frequent bullous dermatosis of children. Dermatol Online J. 2010 15; 16:2. [ Links ]
12. Nantel-Battista M, Al Dhaybi R, Hatami A, Marcoux D, Desroches A, Kokta V. Childhood linear IgA bullous disease induced by trimethoprim-sulfamethoxazole. J Dermatol Case Rep. 2010 19; 4:33-5. [ Links ]
13. Patsatsi A. Chronic Bullous Disease or Linear IgA Dermatosis of Childhood -Revisited. J Genet Syndr Gene Ther 2013; 4:151. [ Links ]
14. Moleiro S, Santos V, Calha M, Pessoa G. Atypical response to treatment in linear IgA bullous dermatosis of childhood: Revision of literature. Dermatol Online J. 2011; 17:5. [ Links ]
15. Horiguchi Y, Ikoma A, Sakai R, Masatsugu A, Ohta M, Hashimoto T. Linear IgA dermatosis: report of an infantile case and analysis of 213 cases in Japan. J Dermatol. 2008; 35:737-43. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Sandra Pereira
Pediatrics Department
Hospital Pediátrico Integrado
Centro Hospitalar Universitário São João
Alameda Prof. Hernâni Monteiro,
4200-319 Porto
Email: sandravdpereira@gmail.com
Received for publication: 06.09.2017
Accepted in revised form: 11.12.2017

