










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.27 no.3 Porto set. 2018
https://doi.org/10.25753/BirthGrowthMJ.v27.i3.12753
CASE REPORTS | CASOS CLÍNICOS
Dark skin - constitutional or pathological? A X-linked Adrenoleukodystrophy case report
Pele escura – constitucional ou patológica? Um caso clinico de Adrenoleucodistrofia ligada ao X
Clara PretoI; José Eduardo AlvesII; Marcelo FonsecaIII; Manuela Santos; Natalina MiguelI
I Department of Pediatrics, Centro Hospitalar de Trás-os-Montes e Alto Douro. 5000-508 Vila Real, Portugal. clarampreto@hotmail.com; natalina.miguel@gmail.com
II Department of Neuroradiology, Centro Hospitalar do Porto. 4099-001 Porto, Portugal. zeedualves@gmail.com
III Department of Pediatric Endocrinology, Hospital Pedro Hispano, Unidade Local de Saúde de Matosinhos. 4460-352 Senhora da Hora, Portugal. mmarcelo.fonseca@gmail.com
IV Department of Neuropediatrics, Centro Materno Infantil do Norte, Centro Hospitalar do Porto. 4099-001 Porto, Portugal. manuela.a.santos@gmail.com
ABSTRACT
Introduction: X-linked adrenoleukodystrophy is a genetically determined peroxisomal disease.
Clinical case: An eleven-year-old boy was referred to a pediatric clinic due to generalized hyperpigmentation beginning at the age of six. By ten years of age he started to present behavior changes and decreased school perfomance. History of cutaneous hyperpigmentation was documented in the boys maternal uncle. Blood tests were compatible with adrenal insufficiency. Brain Magnetic Resonance Imaging showed frontal leukoencephalopathy. The elevated plasmatic concentration of very long-chain fatty acids and the genotype sequencing of ABCD1 gene established the diagnosis of X-linked adrenoleukodystrophy. The boy´s general condition improved with adrenal insufficiency corticoesteroid treatment however progressive cognitive function deterioration was maintained.
Discussion/Conclusion: Early diagnosis and treatment of this rare condition is very important as it can change the disease course. In this case report, given the severity of neurological involvement at diagnosis, no treatment was available to halt neurological disease progression.
Keywords: Adrenal insufficiency; cerebral demyelination; dark skin; X-linked adrenoleukodystrophy
RESUMO
Introdução: A adrenoleucodistrofia ligada ao X é uma doença peroxissomal geneticamente determinada.
Caso clínico: Criança de 11 anos, encaminhada para a consulta de Pediatria por hiperpigmentação generalizada com início aos seis anos de idade. Apresentava comportamento regressivo e diminuição do desempenho escolar desde os dez anos. Documentada história de hiperpigmentação cutânea num tio materno. Os exames laboratoriais foram compatíveis com insuficiência suprarrenal. A Ressonância Magnética Cerebral revelou leucoencefalopatia frontal. A concentração plasmática elevada de ácidos gordos de cadeia muito longa e a sequenciação do gene ABCD1 permitiram a confirmação do diagnóstico de Adrenoleucodistrofia ligada ao X. O estado geral da criança melhorou com o tratamento sintomático; no entanto a deterioração progressiva da função cognitiva manteve-se.
Discussão/Conclusão: O diagnóstico e tratamento precoce desta condição rara é muito importante, uma vez que pode mudar o curso da doença. Neste caso clínico, dada a gravidade do envolvimento neurológico ao diagnóstico, não existe tratamento disponível que seja eficaz na interrupção da progressão da doença neurológica.
Palavras-chave: Adrenoleucodistrofia ligada ao X; desmielinização cerebral; insuficiência supra-renal; pele escura
INTRODUCTION
X-linked adrenoleukodystrophy (X-ALD) is a genetically determined disorder, characterized by accumulation of very-long-chain fatty acids (VLCFA) in plasma, fibroblasts and tissues and progressive demyelination within central and peripheral nervous system, associated with primary adrenocortical insufficiency.1-3
X-ALD results from mutations in the ABCD1 gene, located on the X-chromosome, which encodes a peroxisomal transmembrane protein (ALD protein) member of the ATP-binding cassette transporter superfamily.2 This protein transports VLCFA or their Coenzyme A (CoA) ester derivatives into the peroxisomes, where they are degraded by a peroxisomal beta-oxidation system.1-3 Mutations in this gene results in accumulation of VLCFA in plasma and tissues, mostly in the brain and adrenal cortex, the hallmark of this disease.3
X-ALD is the most common peroxisomal disease.1 The global incidence is 1:17.000 including hemizygotes and heterozygotes both of which frequently symptomatic.1 The phenotype does not correlate with the genotype, suggesting that modifier genes or environmental factors model the clinical outcome of the disease.2
The X-ALD clinical spectrum is very broad and cannot be predicted through levels of VLCFA or family history.4 Three main phenotypes can be distinguished.
Childhood cerebral form is the most devastating and rapidly progressive phenotype, commonly leading to total disability in six months to two years followed by death at varying ages.4 It occurs in 37% of X-ALD cases and primarily affects boys.2,4 Presentation occurs, most frequently, between the ages of four and eight years.4 Typically they present learning disabilities and behavior problems, followed by neurological deterioration that includes increasing cognitive and behavioral abnormalities, blindness, deafness, cerebellar ataxia, seizures or spastic tetraparesis. Brain Magnetic Resonance Imaging (MRI) classically demonstrates cerebral white matter demyelination in occipitoparietal region.5
Adrenomyeloneuropathy usually presents in young adults (second to fourth decade) and comprises approximately 40 to 45% of X-ALD cases.4 The primary manifestation is spinal cord dysfunction with progressive stiffness and weakness of the legs, abnormal sphincter control, and sexual dysfunction.4 Cerebral demyelination can be present in about 40%-45% of cases.4
Isolated adrenal insufficiency (Addison disease only) can occur in 10% of X-ALD cases and may be present in more than 50% of patients with cerebral X-ALD / AMN.6 The signs of high ACTH secretion may include unexplained vomiting, weakness, coma or hyperpigmentation. X-ALD is a frequent cause of Addisons disease (35%), particularly when circulating adrenocortical autoantibodies are absent.1
Phenotypes are not static.1 Non-symptomatic patients are at risk of developing neurologic (cerebral X-ALD, AMN) or endocrinologic (Addisons disease) symptoms. The X-ALD severity and progression cannot be individually predicted.1
The clinical history as well as the findings of physical examination suggests the X-ALD diagnosis. The VLCFA panel is highly sensitive for detecting X-ALD and is the appropriate first step in diagnosis. If the initial screening shows high serum VLCFA levels, or abnormal ratios of VLCFA, ABCD1 gene analysis should be performed to accomplish the diagnosis.1,2
There are few effective therapeutic options for X-ALD patients. Hematopoietic stem cell transplantation (HCT) is the only available therapeutic approach that can stop cerebral demyelination and results in long term quality of life, provided the procedure is performed at an early stage of disease.7
CASE REPORT
An eleven-year-old boy was referred to pediatric clinic care due to generalized hyperpigmentation beginning at the age of six. Until he was ten, no abnormal psycho-motor development or limited academic performance was documented. Since then, concentration and memory deficit, as well as decreased performance at school developed. Additionally, the boy presented hyperkinesia and akathisia. Gastrointestinal complaints like nausea or vomiting were not reported. No other relevant facts of his medical history were noticed. He was the first child of unrelated healthy parents. Mild cutaneous hyperpigmentation was notice in a maternal uncle.
Examination findings revealed adynamia and generalized skin hyperpigmentation, oral mucosa, palmar creases, ungueal beds and nipples. (Figure 1 and 2) Growth parameters were normal for sex and age and the body mass index was in the fifth percentile. His blood pressure was within normal limits. Concerning his sexual development, he was in stage one of Tanner. Neurological examination disclosed disturbed attention, unable to stay still, apraxia and brisk reflexes. There were no other significant findings in the remainder of the clinical examination.
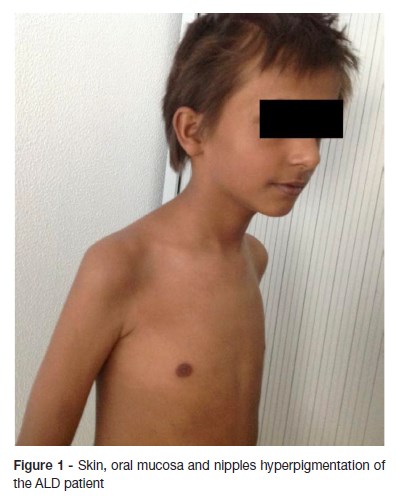
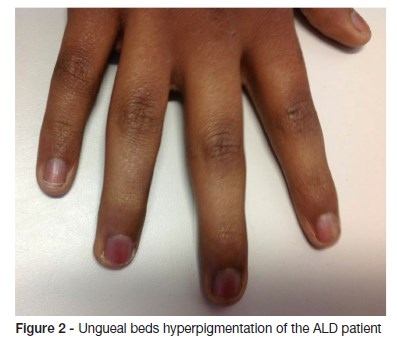
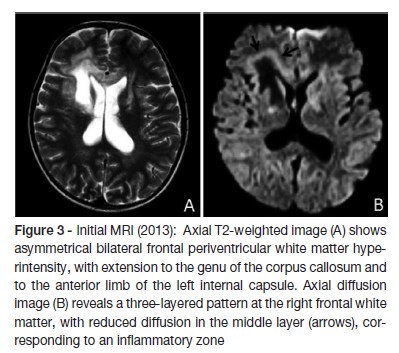
Laboratory results showed blood count, electrolytes (sodium, potassium, calcium), liver and renal function within normal limits. The decreased level of cortisol (1.5 nmol/L) and increased level of adrenocorticotrohin hormone (ACTH - 2000 ng/dL) confirmed adrenal insufficiency. The levels of renin and aldosterone were within normal range. The quantification of very long chain fatty acid showed abnormally high levels, compatible with a peroxisomal beta-oxidation disease: C26:0 0.97 μg/ml (normal range 0.16 - 0.57); C24:0/C22:0 1.62 μg/ml (normal range 0.63 - 1.10); C26:0/C22:0 0.078 μg/ml (normal range 0.004 - 0.022). The biochemical diagnosis was confirmed by molecular analysis: ABCD1 gene sequencing revealed the previously described c.1866-10G>A (p.R622fs*16) pathological variant in hemizygosity. Upper abdomen computed tomography scan showed no structural abnormalities. Brain MRI showed bilateral periventricular frontal white matter T2 hyperintensity (asymmetrical, with right predominance), that extended to genu of the corpus callosum, to the anterior limb, to genu of the internal capsules and to the anterior thalami. Right frontal periventricular white matter involvement displayed a three-layered pattern with an internal zone of high diffusibility and an intermediate rim of reduced diffusion, depicting ongoing inflammation/demyelination. (Figure 3)
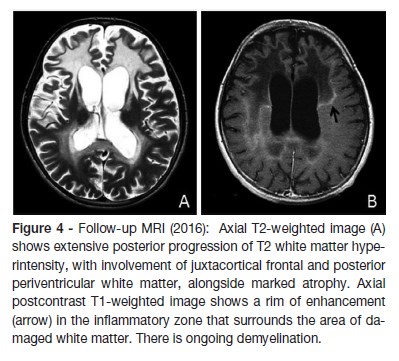
Treatment was started with glucocorticoid replacement (hydrocortisone in increasing doses, maximum of 30mg/m2 and fludrocortisone was added to maximize results), diet therapy with decreased intake of fatty foods, Lorenzo´s oil and physiotherapy. One year later, the Lorenzo´s oil was suspended due to the development of a severe thrombocytopenia. After beginning of this therapeutic approach he had an improvement in his general condition, gait and skin color. Nevertheless he progressively developed dementia and pyramidal syndrome. Three years after the diagnosis, he started seizures, some of them difficult to control, with admission in pediatric intensive care unit due to status epilepticus. Follow-up MRI showed extensive progression of T2 white matter hyperintensity, with involvement of frontal justacortical and posterior periventricular white matter, as well marked atrophy (Figure 4). Four years after the diagnosis, he is unable to fulfill orders, has poor social contact, and restlessness when standing up. He presents language disturbances namely echolalia. Eye contact and visual acuity remains normal and bilateral grasping and sucking are present. Epilepsy is controlled with three antiepileptic drugs.
The family was referred for genetic counseling.
DISCUSSION
X-ALD is a frequent cause of Addisons disease in boys and adult males.8 Adrenocortical insufficiency can be its presenting symptom and occur years before the onset of neurological manifestations.8 The only symptom related to the adrenocortical insufficiency present in this case was the cutaneous darkening from the age of six, but it was overlooked due to family clinical history of cutaneous hyperpigmentation. Symptoms due to low cortisol levels, such as weakness and hypotension, were not referred. Therefore, an early diagnosis of adrenocortical insufficiency was hindered and even masked. Nonspecific neurological presentation is common at the early stage of X-ALD disease and may lead to a wrong and/or delayed diagnosis.4 Typically the initial phase is followed by impaired cognition, behavior, vision, hearing and motor function. In the present case, one year before the diagnosis, learning disabilities and behavior problems were indeed observed, reflecting the early predominant involvement of the frontal lobe. The observation of dark cutaneous and mucosal color associated with neurological symptoms first suggested X-ALD disease, which was supported by high ACTH and low cortisol serum levels, and confirmed by high VLCFA levels in plasma and a pathogenic mutation in the ABCD1 gene. The changes in MRI confirmed the cerebral involvement. Most patients with cerebral X-ALD present posterior predominant periventricular white matter demyelinating lesions, extending to the splenium of corpus callosum.5 Nevertheless, in 20% of the cases, initial demyelization occurs in the genu of corpus callosum and then progresses symmetrically or asymmetrically to the periventricular frontal white matter, as occurred in this case.5
Treatment was based on corticosteroid replacement therapy targeted to adrenal insufficiency, despite its lack of effect on the neurological abnormalities in X-ALD cases.9
Hematopoietic stem cell transplantation (HCT) is the only available treatment that can arrest cerebral demyelination of X-ALD in boys.7 Best outcomes are obtained in patients whose HCT is performed in an early stage of the disease and from a related donor.7 The ideal candidates for HCT are males with mild or no neurological deficits and evidence of MRI cerebral involvement presented early in their disease course.4 HCT is not recommended for boys without MRI evidence of cerebral involvement, since many of them will remain free of neurological disease, or in boys with advanced neurological disease, given the scarce evidence of clinical improvement in these patients and the significant morbidity and mortality of this procedure.4 Only 30% of patients who might benefit from a HCT will have a full human leukocyte antigene (HLA) matched donor that is considered to be the best choice. For patients that have no full HLA matched donor and need immediate treatment, transplantation of haploidentical stem cells combined with the infusion of umbilical cord blood can be an option.10 The severity of neurological impairment and the neuroimaging assessment presented in our patient was not in accordance with HCT recommendations.
Early results of an ongoing, multicenter, phase 2-3 study, suggests that hematopoietic stem cell gene therapy with a lentiviral vector is a safe and effective alternative to allogeneic HCT in an early stage of cerebral X-ALD.11
Statins can reduce VLCFA level but have no influence in neuronal and endocrine functions.12 This therapeutic approach was not considered for our patient.
Oral administration of Lorenzos oil, a 4:1 mixture of glyceryl trioleate and glyceryl trierucate, plus moderate reduction of fat in the diet, can normalize the VLCFA levels in plasma, however its clinical efficacy and clinical indications for its use have been controversial for many years. Several studies concluded that there is no clinically relevant benefit from dietary treatment with Lorenzos oil in both asymptomatic and symptomatic patients.13,14 However subsequently, Moser and colleagues demonstrated in a cohort of 89 patients that Lorenzos oil blunts the progress of X-ALD, but only if it is begun before the onset of either MRI changes or neurological manifestations.15 Due to the adverse effects of Lorenzos oil, including thrombocytopenia and elevated liver enzymes, its use requires a careful laboratory monitoring. In this case report, although questionable, given the lack of therapeutic options and the expected poor prognosis, a therapeutic trial was carried out being only interrupted one year later due to development of moderate thrombocytopenia.
Corticosteroid replacement improved the boy´s strength, his well being and decreased the hyperpigmentation although, as expected, it had no effect on cerebral disease. Given the severity of neurological involvement at diagnosis, there was no treatment that could stop and reverse his neurological disease progression. Therefore, the child progressed to a dementia status and developed seizures.
We aim, in this report, to highlight the importance of an earlier diagnosis of this rare disease, not only due to its severe morbidity and mortality, but also because early treatment of this disorder can prevent its neurological progression. It is therefore important to consider X-ALD in all the boys with Addisons disease, especially if accompanied by neurological symptoms.
REFERENCES
1. Engelen M, Kemp S, Visser M et al. Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet Journal of Rare Diseases 2012; 7:51-64. [ Links ]
2. Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Appl Clin Genet 2015; 8:109-21. [ Links ]
3. Brett EM, Auchus RJ. Genetic forms of adrenal insufficiency. Endocr Pract 2015; 21:395-9. [ Links ]
4. Moser HW, Moser AB, Steinberg SJ. X-linked adrenoleukodystrophy. GeneReviews. www.genetests.org. (Last Update 09, 2015). [ Links ]
5. Van der Knaap MS, Valk J: X-linked adrenoleukodystrophy. In: Magnetic Resonance of Myelination and Myelin Disorders. Heilmann U (3rd edition). Berlin-Heidelberg-New York: Springer; 2005:176-90. [ Links ]
6. Ronghe MD, Barton J, Jardine PE et al. The importance of testing for adrenoleucodystrophy in males with idiopathic Addisons disease. Arch Dis Child 2002; 86:185-9. [ Links ]
7. Saute JA, Souza CF, Poswar FO, Donis KC, Campos LG, Deyl AV, et al. Neurological outcomes after hematopoietic stem cell transplantation for cerebral X-linked adrenoleukodystrophy, late onset metachromatic leukodystrophy and Hurler syndrome. Arq Neuropsiquiatr 2016; 74:953-66. [ Links ]
8. Laureti S, Casucci G, Santeusanio F, Angeletti G, Aubourg P, Brunetti P. X-linked adrenoleukodystrophy is a frequent cause of idiopathic Addisons disease in young adult male patients. J Clin Endocrinol Metab 1996; 81:470-4. [ Links ]
9. Berger J, Pujol A, Aubourg P, Forss-Petter S. Current and Future Pharmacological Treatment Strategies in X Linked Adrenoleukodystrophy. Brain Pathol. 2010; 20:845-56. [ Links ]
10. Jiang H, Jiang MY, Liu S, Cai YN, Liang CL, Liu L. Combination of a Haploidentical Stem Cell Transplant With Umbilical Cord Blood for Cerebral X-Linked Adrenoleukodystrophy. Pediatr Neurol 2015; 53:163-5. [ Links ]
11. Eichler F, Duncan C, Musolino PL, Orchard PJ, Oliveira S, Thrasher AJ et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N Engl J Med 2017; 377: 1630-8. [ Links ]
12. Engelen M, Ofman R, Dijkgraaf MG, et al. Lovastatin in X-linked adrenoleukodystrophy. N Engl J Med 2010; 362:276-7. [ Links ]
13. Aubourg P, Adamsbaum C, Lavallard-Rousseau MC, Rocchiccioli F, Cartier N, Jambaqué I, et al. A two-year trial of oleic and erucic acids (Lorenzos oil) as treatment for adrenomyeloneuropathy. N Engl J Med 1993; 329:745-52. [ Links ]
14. van Geel BM, Assies J, Haverkort EB, Koelman JH, Verbeeten B Jr, Wanders RJ, et al. Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X linked adrenoleukodystrophy despite treatment with Lorenzos oil. J Neurol Neurosurg Psychiatry 1999; 67:290-9. [ Links ]
15. Moser HW, Raymond GV, Lu SE, Muenz LR, Moser AB, Xu J, et al. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzos oil. Arch Neurol 2005; 62:1073-80. [ Links ]
CORRESPONDENCE TO
Clara Preto
Department of Pediatrics
Centro Hospitalar de Trás-os-Montes e Alto Douro
Avenida Noruega
5000-508 Vila Real
Email: clarampreto@hotmail.com
Received for publication: 06.08.2017
Accepted in revised form: 20.11.2017

