








Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkNascer e Crescer
Print version ISSN 0872-0754On-line version ISSN 2183-9417
Nascer e Crescer vol.27 no.3 Porto Sept. 2018
https://doi.org/10.25753/BirthGrowthMJ.v27.i3.11686
CASE REPORTS | CASOS CLÍNICOS
Trisomy 20 mosaicism – a subtle phenotype
Trissomia 20 em mosaico – um fenótipo subtil
Inês MedeirosI; Catarina FariaI; Fábia CarvalhoI; Miguel Gonçalves RochaII; Helena SilvaI
IDepartment of Pediatrics, Hospital de Braga. 4710-243 Braga, Portugal. inesdemedeiros@hotmail.com; catmagalhaesfaria@gmail.com; fsgc_carvalho@hotmail.com; tizleite@hortmail.com
IIDepartment of Genetics, Hospital de Braga. 4710-243 Braga, Portugal. miguel.rocha@hospitaldebraga.pt
ABSTRACT
Introduction: Trisomy 20 mosaicism is one of the most common cytogenetic abnormalities found in prenatal diagnosis. The outcome of these pregnancies is normal in most of the reported cases, but clinical implications of diagnosis and prognosis in a long term basis are not clear.
Case report: The authors present a case of trisomy 20 mosaicism, with no prenatal diagnosis, followed-up for a period of 13 years, aiming to demonstrate the existence of a subtle phenotype, which represents a challenge in diagnosis.
Discussion/Conclusion: No specific phenotype has been associated with the cytogenetic findings but certain features, although subtle, appear to be consistent.
Keywords: Mosaicism; phenotype; trisomy 20
RESUMO
Introdução: A trissomia 20 em mosaico é um dos tipos mais comuns de alterações citogenéticas detetadas no diagnóstico pré-natal. O produto destas gestações é normal na maioria dos casos descritos, mas as implicações clínicas do diagnóstico e o prognóstico da doença a longo prazo não são totalmente claros.
Caso clínico: Os autores apresentam um caso de trissomia 20 em mosaico, sem diagnóstico pré-natal e com um período de seguimento de 13 anos, pretendendo ilustrar a existência de um fenótipo subtil, o que constitui um desafio no diagnóstico.
Discussão/Conclusão: Nenhum fenótipo específico tem sido associado com os achados citogenéticos, mas certas características apesar de subtis parecem ser consistentes.
Palavras-chave: fenótipo; mosaicismo; trissomia 20
INTRODUCTION
Despite being a frequently found cytogenetic abnormality in amniocentesis and chorionic villus sampling (1 out of 7000 pregnancies), clinical trisomy 20 mosaicism is rarely identified in the postnatal period.1,2 In this condition, an extra copy of chromosome 20 (or a portion of it) is found in some cell lines. The outcome of these pregnancies is normal in 90-93% of cases with prenatal diagnosis; however, most studies only consider clinical features at birth and have a short follow-up period. Non-mosaic trisomy 20 is, in most cases, incompatible with life.1,3
Clinical findings at birth include hypotonia and skin pigmentation; later, delayed psychomotor development may be noticed. Other findings include vertebral column abnormalities (vertebral fusion and kyphosis), narrowed chest and sloping shoulders, congenital heart and kidney defects, visual alterations and learning difficulties.2,4,8,9
Cytogenetic analysis of skin fibroblasts is the most appropriate technique for postnatal confirmation of the diagnosis, as most trisomy 20 mosaicism cases have a normal pheripheral blood karyotype.10
CASE REPORT
The authors present a thirteen-year-old female, born at 39 weeks by eutocic delivery. Third-trimester serologies were unremarkable. Fetal ultrasound detected a bilateral hydronephrosis and reduced left renal parenchyma. Family history was irrelevant with no parental consanguinity.
Hypotonia and multiple equimosis-like lesions in the lumbar region and limbs were noticed at birth. Coagulation study was normal and renal-pelvic ultrasound performed during the first week of life revealed left renal agenesis and right hydronephrosis.
At five months, the infant developed bilateral convergent strabismus. The equimosis-like lesions gave place to cutaneous lesions similar to mongolian spots of atypical configuration (Figure 1). Hypotonia was sustained. Transfontanelar ultrasound and cranioencephalic magnetic resonance imaging (MRI) excluded a neurocutaneous syndrome.
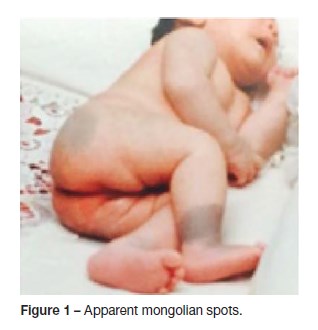
At nine months, a delay in psychomotor development was noticed. Peripheral blood karyotype, observed in 10 cells, was 46,XX; metabolic study and auditory evoked potential were unremarkable. She maintained bilateral convergent strabismus and initiate ocular occlusion.
A scoliotic position and changes in gait were noted at the age of 20 months. Vertebral magnetic resonance imaging confirmed severe dorsal scoliosis and revealed anterior fusion of the first two ribs. A heart murmur was detected and the echocardiogram identified a bicuspid aortic valve, without regurgitation or stenosis.
Between the age of three and six years, speech and learning disabilities were detected. The child started speech therapy and was engaged into special pedagogic support. At six years, she repeated cranioencephalic MRI, which showed no alterations.
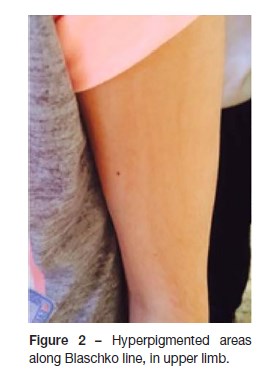
At the age of nine, the apparent mongolian-like spots became progressively clearer and linear areas of hyperpigmentation along Blaschko lines (Figure 2). Skin biopsy karyotype was performed and two distinct cell lines were identified: the first one with 47 chromosomes and an extra chromosome 20 and the other one with 46 chromosomes, of normal constitution (47,XX,+20[23]/46,XX[27]).
Currently, at the age of thirteen years, the adolescent presents low cognitive functioning (Intelligence Quotient 71) according to the Wechsler Intelligence Scale for Children III, maintaining the need for individualized pedagogical support at school. She presents a mild strabismus even after several correction surgeries and needs to wear glasses, a narrow chest and sloping shoulders (Figure 3), mild hypotonia and fine motor skills disabilities and is under physiatric treatment. She has a multidisciplinary follow-up (Pediatrics, Physical and Rehabilitation Medicine, Ophthalmology, Orthopedics and Cardiology) and surgical correction of scoliosis is planned.

DISCUSSION/CONCLUSION
There are few clinical cases of trisomy 20 mosaicism reported in the literature. The number of prenatally detected cases is higher than the true number of children with pathology, so it is suspected that many trisomy cells are restricted to the placenta.4 It is suggested that a higher percentage of affected cells in the prenatal period increases the risk of an abnormal outcome; however, this correlation is not considered significant for genetic counseling.5,6,7
Although there is no specific phenotype or syndrome described, certain clinical features appear to be recurrent, including skin pigmentation alterations, hypotonia and delayed psychomotor development.1,2,4 Long-term follow-up is required, as sometimes orthopedic, ophthalmologic, cardiac and renal features may arise.
The authors underline the fact that the exclusion of mosaicism implies counting a minimum of 30 cells. The peripheral blood karyotype did not excluded mosaicism reliably, once only 10 cells were observed, but the skin karyotype confirmed our clinical suspicion.
Our case sets forth the challenge of diagnosis in the absence of prenatal finding of cytogenetic abnormalities. As the clinical implications and prognosis of this pathology are not yet clear the authors consider important to accurately characterize all detected cases and follow them closely as well as providing genetic counselling for the parents.
REFERENCES
1. Hsu L, Kaffe S, Perlis T. A revisit of trisomy 20 mosaicism in prenatal diagnosis – an overview of 103 cases. Prenat Diagn. 1991; 1:7-15. [ Links ]
2. Bui T-H, Iselius L, Lindsten J. European collaborative study on prenatal diagnosis: Mosaicism, pseudomosaicism and single abnormal cells in amniotic fluid cell cultures. Prenat Diagn. 1984; 4:145-62. [ Links ]
3. Beke A, Tóth-Pál E, Hargitai B, Szigeti Z, Papp C, Papp Z. Trisomy 20 mosaicism and nonmosaic trisomy 20: a report of 2 cases. J Reprod Med. 2006; 51:209-12. [ Links ]
4. Willis MJ, Bird LM, DellAquilla M, Jones MC. Expanding the Phenotype of Mosaic Trisomy 20. Am J Med Genet A. 2008; 146A:330-6. [ Links ]
5. Robinson WP, McGillivray B, Lewis ME, Arbour L, Barrett I, Kalousek DK. Prenatally detected trisomy 20 mosaicism. Prenatal Diagnosis. 2005; 25:239-44. [ Links ]
6. Chen CP, Chang SD, Chueh HY. Discrepancy in the trisomy mosaicism level between cultured amniocytes and uncultured amniocytes in prenatally detected mosaic trisomy 20. Taiwan J Obstet Gynecol. 2013;145-6. [ Links ]
7. Bianca S, Boemi G, Barrano B, Cataliotti A, Ingegnosi C, Indaco L, et al. Mosaic trisomy 20: considerations for genetic counseling. Am J Med Genet. 2008; 146A:1897-8. [ Links ]
8. Baty BJ, Olson SB, Magenis RE, Carey JC. Trissomy 20 mosaicism in two unrelated girls with skin hypopigmentation and normal intellectual development. Am J Med Genet. 2001; 99:210-6. [ Links ]
9. Hartmann A, Hofmann UB, Hoehn H, Broecker EB, Hamm H. Postnatal Confirmation of Prenatally Diagnosed Trisomy 20 Mosaicism in a Patient with Linear and Whorled Nevoid Hypermelanosis. Ped Dermatology. 2004; 21:636-41. [ Links ]
10. Taibjee SM, Hall D, Balderson D, Larkins S, Stubbs T, Moss C. Keratinocyte cytogenetics in 10 patients with pigmentary mosaicism: identification of one case of trisomy 20 mosaicism confined to keranocytes. Clin Exp Dermatol. 2009;34:823-9. [ Links ]
Inês Medeiros
Department of Pediatrics
Hospital de Braga
Rua das Sete Fontes
4710-243 Braga
Email: inesdemedeiros@hotmail.com
Received for publication: 05.03.2017
Accepted in revised form: 02.10.2017