










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.24 no.2 Porto jun. 2015
CASOS CLÍNICOS | CASE REPORTS
Policitemia vera: a propósito de um caso clínico
Polycythemia vera: a case report
Joana MacedoI; Emília CostaII; José BarbotII ; Cláudia NetoI
IS. Pediatria, Centro Hospitalar do Alto Ave, 4180-052 Guimarães, Portugal. E-mail: macedojoaninha@hotmail.com; claudianeto76@gmail.com
IIUnidade de Hematologia Pediátrica, Centro Hospitalar do Porto, 4099-001 Porto, Portugal. E-mail: emvcosta@gmail.com; josebmbarbot@gmail.com
RESUMO
A policitemia vera (PV) é um transtorno mieloproliferativo das células hematopoiéticas, caracterizada por uma produção anormal e acentuada de eritrócitos, leucócitos e plaquetas. Outras características da doença incluem esplenomegalia, complicações trombo-hemorrágicas, distúrbios vasomotores, prurido e um pequeno risco de progressão da doença para leucemia mielóide aguda ou mielofibrose. A trombose é o sintoma de apresentação em 20% dos pacientes com PV. É uma doença rara, com uma incidência de 2,3/100.000 pessoas por ano, que é ainda mais incomum em crianças e adolescentes. É obrigatório o diagnóstico diferencial com as outras doenças mieloproliferativas.
Caso clínico: Descreve-se o caso clínico de uma adolescente do sexo feminino, 14 anos de idade, referenciada à consulta de hematologia pediátrica por trombocitose. Os achados clínicos e analíticos sugestivos de PV foram confirmados após diagnóstico molecular. Iniciou terapêutica com alfa-interferão 2a. Atualmente apresenta contagem plaquetária e eritrocitária dentro de valores normais.
Conclusões: Os autores fazem uma análise do caso clinico, inserido no contexto mandatório de um diagnóstico diferencial entre trombocitose reativa e outras doenças mieloproliferativas.
Palavras-chave: Adolescência; Policitemia vera; Trombose; Trombocitose
ABSTRACT
Polycythemia vera (PV) is a myeloproliferative disturbance of haematopoietic cells characterized by abnormal and overstated production of erythrocytes, leukocytes and platelets. Other disease features include splenomegaly, thrombohemorrhagic complications, vasomotor disturbances, pruritus and a small risk of disease progression into acute myeloid leukemia or myelofibrosis. Thrombosis is the presenting symptom in 20% of patients with PV. It is a rare disease with an incidence of 2.3/100.000 people per year, and is even more uncommon in children and adolescents.
We present a case report of a fourteen-year-old years old adolescent with clinical and laboratorial findings suggestive of polycythemia vera. Treatment with alpha-interferon was initiated. Erythrocyte and platelet count are now in the normal range.
The authors make, in the context of this case report, a brief review of the criteria for the differential diagnosis of reactive thrombocytosis and myeloproliferative diseases, manifestations and treatment options.
Keywords: Adolescence; Polycythemia vera; Thrombosis; Thrombocytosis
INTRODUÇÃO
As doenças mieloproliferativas (Myeloproliferative diseases (MPD)) constituem um grupo heterogéneo de patologias, onde ocorre produção excessiva de um ou mais dos elementos sanguíneos sem displasia significativa e em que há tendência para hematopoiese extramedular, mielofibrose e transformação para leucemia aguda. De acordo com o sistema de classificação da Organização Mundial de Saúde (OMS) para doenças hematopoiéticas, a policitemia vera (PV), pertence à subcategoriaBCR-ABL1-negativo. A principal característica fisiopatológica é a hiperplasia das células hematopoiéticas, independente da produção de eritropoietina (EPO), levando a uma acentuada produção de eritrócitos, leucócitos e plaquetas.1 O diagnóstico de PV baseia-se atualmente nos critérios da OMS, partindo de uma avaliação composta de diversas caraterísticas clínicas e laboratoriais.
Considerada uma doença rara, a sua incidência é de 2,3/100.000 pessoas por ano.2,5 Embora possa ocorrer em qualquer faixa etária, a idade média dos pacientes é de 60 anos, com predomínio do sexo masculino.1,2,5 Apenas 1% dos pacientes apresentam a patologia antes dos 25 anos de idade, e apenas 0,1% têm na altura do diagnóstico, menos de 20 anos. Portanto, até o momento, são raros os casos descritos de PV na faixa etária da pediatria.
A sua heterogeneidade fenotípica, que dificulta o diagnóstico diferencial, tem uma base genética que se deve à ativação constitucional de vias de transdução de sinais, causadas por rearranjos genéticos ou mutações que afetam as proteínas tirosina cinases, como a JAK2, ou moléculas relacionadas.
De acordo com as orientações da OMS, a PV deve ser suspeitada em doentes com níveis de hemoglobina superiores a 18,5}g/dL nos homens ou 16,5g/dL nas mulheres ou níveis de hemoglobina superiores a 17g/dL nos homens ou a 15g/dL nas mulheres se associados a um aumento sustentado de pelo menos 2 g/dL relativamente à linha de base individual. Outro critério major no diagnóstico da PV á a presença de mutação na JAK2.
Critérios minor de diagnóstico são o nível sérico baixo de eritropoietina (EPO), histologia da medula óssea compatível com MPD e o crescimento de colónias eritróides endógenas. O diagnóstico de PV requer a presença de ambos os critérios major e um critério minor ou o primeiro critério major e dois critérios minor.
Relativamente à apresentação clínica da patologia, tendo em conta que a eritrocitose causa hiperviscosidade, pode cursar com sintomas neurológicos como vertigem, cefaleias, altera- ções visuais e acidentes isquémicos transitórios. A hipertensão sistólica é também uma característica da elevação da massa eritrocitária. A esplenomegalia pode ser o sinal de apresentação inicial da PV, mas esta é detetada mais frequentemente pela descoberta incidental de valores elevados de hemoglobina ou hematócrito.
As principais complicações da PV também estão diretamente relacionadas com o aumento da viscosidade sanguínea, associada à elevação da massa eritrocitária, leucocitária e plaquetar. Em alguns doentes, a trombose venosa ou arterial pode ser a manifestação de apresentação. A trombose venosa intra-abdominal é particularmente comum em mulheres jovens e pode ser catastrófica se ocorrer uma obstrução súbita e completa da veia hepática. O desenvolvimento de leucemia está relacionado com a idade mais avançada, mas não com a duração da doença, sugerindo que a exposição ao tratamento será um fator de risco mais importante do que a doença em si.1,3 A maioria dos eventos trombóticos ocorre nos dois primeiros anos a seguir ao diagnóstico.2,4
O objetivo da intervenção terapêutica atual na PV é a evicção de complicações trombo-hemorrágicas, sem aumentar o risco de hemorragia e, secundariamente, o controlo dos sintomas acima referidos. Desta forma, perante a ausência de consensos, o tratamento é adaptado individualmente a cada doente, baseado na clínica e forma de apresentação da patologia, sobretudo de acordo com o seu risco de trombose ou de hemorragia.
A sobrevida média dos pacientes sintomáticos sem tratamento é de 6-18 meses, enquanto que a daqueles com suporte adequado pode ser maior de 10 anos.7 A taxa de mortalidade é dependente da idade, sendo 1,6 vezes ou 3,3 vezes superior à população de referência, em doentes com menos ou mais de 50 anos, respetivamente.8
CASO CLÍNICO
Os autores descrevem o caso clínico de uma adolescente do sexo feminino, de 14 anos de idade com antecedentes pessoais e familiares irrelevantes. Referenciada à consulta de pediatria de doenças hematológicas, devido a apresentar laboratorialmente uma trombocitose sustentada (contagem de plaquetas entre 713.0001.107.000 / mm3) com um ano de evolução. Assintomática até cerca de um ano antes da respetiva consulta de doenças hematológicas, tendo iniciado nessa altura um quadro de cefaleias recorrentes, com localização frontal bilateralmente, ocorrendo com predomínio vespertino e acompanhadas de fenómenos visuais prodrómicos (escotoma cintilante e fotofobia). Negava quaisquer outros sintomas, tais como emagrecimento, febre, sudorese noturna, ingestão de medicamentos e doenças ou cirurgias prévias.
Apresentava bom estado geral e de nutrição, sem alterações cutâneas, exame neurológico sem défices e o restante exame físico não revelou quaisquer alterações.
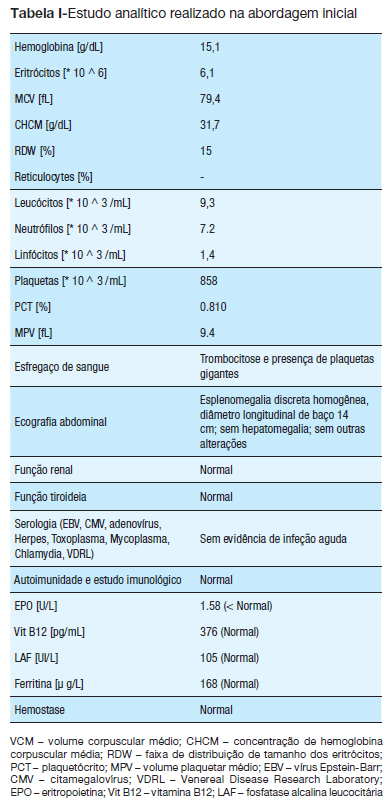
Dos exames complementares realizados, além de confirmada trombocitose (valor inicial: 858.000/u1), mostraram hemoglobina:15,1g/dL, hematócrito: 47,7% e leucócitos: 9.300/dL (com 52% de neutrófilos e 38% de linfócitos). Ferritina, sideremia, proteína C reativa, velocidade de sedimentação, desidrogenase láctica, ácido úrico, fosfatase alcalina leucocitária, tempo de protombina, tempo de tromboplastina parcial ativado e fibrinogénio revelaram-se dentro dos valores normais (Tabela I). A ecografia abdominal mostrou um baço com textura homogénea, discretamente aumentado (cerca de 14cm). Foram realizadas várias serologias víricas, bem como estudo de imunidade, que não revelaram qualquer alteração. O esfregaço de sangue periférico mostrou plaquetas aumentadas de tamanho.
Devido à trombocitose extrema e persistente sem causa identificada, após exclusão da maioria das causas mais frequentes de trombocitose reativa, decide-se realizar punção medular aspirativa (PMA) e biópsia óssea (BO). Na PMA a análise citomorfológica evidenciou hipercelularidade medular com proliferação megacariocítica proeminente. A BO foi compatível com patologia mieloproliferativa: O exame histológico mostra um síndrome mieloproliferativo com trombocitose marcada.
Entretanto, no decorrer do seguimento, devido ao aumento da intensidade e gravidade das cefaleias foi realizada uma angiografia cerebral, tendo sido diagnosticado trombose do segmento posterior do seio longitudinal e trombose do segmento superior do seio lateral esquerdo.
Com a exclusão de grande parte das causas de trombocitose reativa e a apresentação clínica de um evento trombótico, vieram corroborar a existência etiológica de uma patologia mieloproliferativa ou distúrbio mielodisplásico, que precisava de ser especificado. O estudo laboratorial complementar, que entretanto estava a ser realizado, revelou um aumento da massa eritrocitária (> 39,6% acima do valor médio normal); o valor de EPO sérica estava baixo (1.58U/l – faixa normal: 3.7-19.4U/l); A fosfatase alcalina leucocitária apresentava um valor normal de acordo com os valores de referência para a idade ((94 (intervalo normal: 20-180)); o doseamento da vitamina B12 estava normal, ((376pg / ml (intervalo normal, 200-835)) e, finalmente, a presença da mutação de JAK2 V617F foi positiva. Com base nestes resultados, foi possível confirmar o diagnóstico de PV.
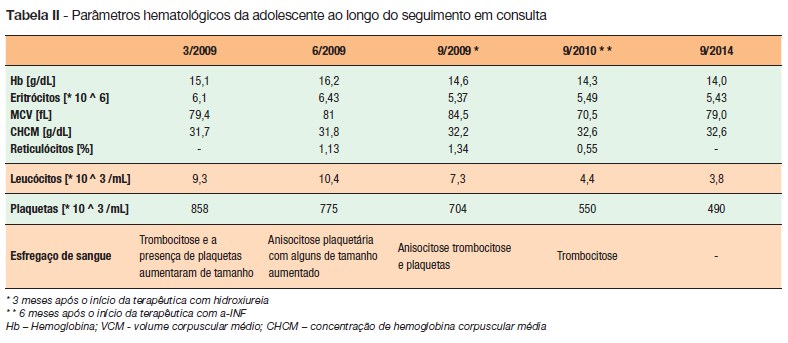
Iniciou terapêutica mielossupressiva com hidroxiureia (500mg/dia) e antiplaquetária com aspirina em baixa dose (100mg/dia). Três meses depois, a adolescente estava assintomática, mas mantinha trombocitose sustentada (contagem de plaquetas entre 730 a 858x109/L), um nível de hemoglobina entre 15,7-14,6g/dL, um hematócrito elevado (41 a 45,7%), com manutenção de leucograma normal (leucócitos 8.800 a 10,200/dL) (Tabela II). Após discussão clínica com Hematologistas Pediátricos, decidiu-se iniciar o tratamento com alfa-Interferão 2a (a-IFN), 3.000.000UI, administração subcutânea 3 vezes por semana. Seis meses após a mudança terapêutica, apesar de manter níveis semelhantes de hematócrito, teve uma redução franca do número de plaquetas (Tabela II).
Atualmente, com mais de três anos de tratamento com a-IFN, a adolescente está assintomática, sem tendência hemorrágica ou trombótica e sem outras complicações. Mantém a regressão hematológica sem critérios de trombocitose e sem eritrocitose: contagem de plaquetas entre 300 a 500x109/L, hemoglobina entre 13 a 14.2g/dL e hematócrito de inferior a 45% (Tabela II).
DISCUSSÃO
A trombocitose, nomeadamente a trombocitose reativa (TR), é um achado comum em Pediatria.7,8 É na maioria dos casos secundária a uma causa bem definida, como infeção, anemia, hipoxemia, inflamação, malignidade, stress e esplenectomia, sendo limitada à resolução da etiologia de base. A sintomatologia depende da condição de base. Como na TR é previsível a descida do número de plaquetas e as complicações trombóticas e/ou hemorrágicas são raras, geralmente não está indicado qualquer tratamento com anticoagulantes ou anti-agregantes plaquetários.1,2,5 Ao contrário da TR, as doenças mieloproliferativas são raras e o diagnóstico diferencial é essencial para uma intervenção precoce.
A PV é causada pela expansão clonal anormal de progenitores eritróides capazes de proliferar na ausência de eritropoietina, mas que cursa em grande parte dos casos com trombocitose grave, que pode, numa primeira fase, atrasar o diagnóstico definitivo. A PV, além de rara, reveste-se de uma particular gravidade nas crianças e adolescentes, tendo em conta que nestes a apresentação clínica e hematológica é mais heterogênea. É mandatória a exclusão de causas secundárias de policitemia, sendo que nestes casos a saturação do oxigénio arterial é normal e é evidente uma esplenomegalia, que também pode ser a forma de apresentação clínica. Na ausência de esplenomegalia existe geralmente leucocitose, trombocitose e concentração de eritropoietina plasmática baixa (<4 mU/mL).16
O diagnóstico da PV é atualmente realizada de acordo com os critérios da OMS e com base numa avaliação composta de características clínicas e laboratoriais.1,2 A deteção da mutação na tirosina cinase (JAK2V617F) é altamente sensível (sensibilida- de de 97%) e específica (especificidade de 100%) para distinguir PV de outras causas de aumento do hematócrito.
A morbilidade da patologia em questão depende sobretudo do risco de trombose e da possibilidade da doença progredir para leucemia aguda ou mielofibrose com metaplasia mielóide (MMM). Segundo a OMS, o risco em 10 anos de transformação leucémica e progressão fibrótica é <5% e <10%, respetivamente, mas o risco de trombose é superior a 20%. Dessa forma a estratificação do risco do doente com PV deve ser realizada com base na probabilidade de haver complicações trombóticas e não necessariamente no risco de transformação fibrótica e/ou leucémica.3, 4,6
Estudos controlados têm confirmado o valor anti-trombótico do uso do ácido acetilsalicílico em baixa dose (40-100mg/dia) em todos os doentes com diagnóstico de PV, mesmo que assintomáticos.1-3,5 Este fármaco também demonstrou ser eficaz no alívio de sintomas vasomotores (microvasculares), perturbações associadas frequentemente com PV.1,5,8 Além do ácido acetilsalicílico, os pacientes de alto risco com PV devem receber agentes citoredutores como hidroxiureia, anagrelide, a-IFN ou busulfan.
No caso apresentado pelos autores, com um evento trombótico a preceder o diagnóstico de PV em uma paciente jovem, a terapêutica citorredutora foi iniciada desde o diagnóstico. Pela ausência de resposta, baseada na inexistência de uma remissão hematológica, esta terapia foi ajustada, com o uso de a-IFN, tendo-se obtido uma boa resposta clínica e laboratorial.
Está atualmente bem eselecido que o a-IFN pode controlar a eritrocitose ou trombocitose na maioria dos pacientes com PV (dose usual é de três milhões de unidades SC três vezes por semana).1 Um grau semelhante de benefício é apreciado em termos de redução do tamanho do baço ou alívio do prurido. Dois estudos recentes sobre o uso de a-IFN em doentes com PV, relataram remissões hematológicas superiores a 80%, acompanhado em alguns casos de remissão molecular completa (5-10%).1 O seu custo elevado, a administração subcutânea e os efeitos colaterais poderão eventualmente limitar o seu uso.6,7 O aparecimento de anticorpos neutralizantes, em 10 a 20% dos casos, pode vir a diminuir a sua eficácia.6 Estudos controlados são necessários para esclarecer a vantagem ou desvantagem do uso de a-IFN em comparação com outros agentes citoredutores, nomeadamente a hidroxiureia.
Conclusão: Uma vez que atualmente não é possível tratar um paciente pediátrico com PV baseado em evidências, é necessária uma estreita cooperação a nível mundial entre médicos e instituições, a fim de otimizar a abordagem a estes doentes.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Tefferi, A., Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, riskstratification, and management. Am. J. Hematol., 88: 507–16. doi: 10.1002/ajh.23417. [ Links ]
2. Vardiman JW, Thiele J, Arber DA, et al. The 2003 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009;114:937-51. [ Links ]
3. Cario H, McMullin MF, Pahl HL. Clinical and hematological presentation of children and adolescents with polycythemia vera. Ann Hematol. 2009;88:713-9. [ Links ]
4. Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 2008;22:14-22. [ Links ]
5. McMullin MF. Idiopathic erythrocytosis: a disappearing entity. Hematology Am Soc Hematol Educ Program. 2009;1:629-35. [ Links ]
6. Percy MJ, Rumi E. Genetic origins and clinical phenotype of familial and acquired erythrocytosis and thrombocytosis. Am J Hematol. 2009;84:46-54. [ Links ]
7. Gordeuk VR, Stockton DW, Prchal JT. Congenital polycythemias/erythrocytoses. Haematologica. 2005; 90:109-16. [ Links ]
8. Osgood EE. Polycythemia vera: age relationship and survival. Blood. 1965; 26:243-56. [ Links ]
9. Wick H. Polycythaemia vera mit neurologischen Komplikationen bei einem 12jährigen Kind. Schweiz Med Wochenschr. 1969; 99:186-9. [ Links ]
10. Park MJ, Shimada A, Asada H, Koike K, Tsuchida M, Hayashi Y. JAK2 mutation in a boy with polycythemia vera, but not in other pediatric hematologic disorders. Leukemia. 2006; 20:1453-4. [ Links ]
11. Aggeler PM, Pollycove M, Hoag S, Donald WG, Lawrence JH. Polycythemia vera in childhood. Studies of iron kinetics with Fe59 and blood clotting factors. Blood. 1961; 17:345-50. [ Links ]
12. Berbis P, Devaux J, Benveniste MJ, Perrimond H, Privat Y. Severe erosive lichen planus and polycythemia vera in an adolescent. Dermatologica. 1987; 174:244-8. [ Links ]
13. Cap J. Polycythemia vera in an 11-year-old child. Bone marrow depression after daraprim treatment. Cesk Pediatr. 1961; 16:49-53. [ Links ]
14. Rives S, Pahl HL, Florensa L, et al. Molecular genetic analyses in familial and sporadic congenital primary erythrocytosis. Haematologica. 2007;92:674-7. [ Links ]
15. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. New Engl J Med 2013;368:22-33. [ Links ]
16. Ang SO, Chen H, Gordeuk VR, et al. Endemic polycythemia in Russia: mutation in the VHL gene. Blood Cells Mol Dis. 2002;28:57-62. [ Links ]
17. Perrotta S, Nobili B, Ferraro M, et al. Von Hippel-Lindaudependent polycythemia is endemic on the island of Ischia: identification of a novel cluster. Blood. 2006;107:514-9. [ Links ]
18. Cario H, Schwarz K, Jorch N, et al. Mutations in the von Hippel-Lindau (VHL) tumor suppressor gene and VHLhaplotype analysis in patients with presumable congenital erythrocytosis. Haematologica. 2005;90:19-24. [ Links ]
19. van Wijk R, Sutherland S, Van Wesel AC, et al. Erythrocytosis associated with a novel missense mutation in the HIF2A gene. Haematologica. 2010;95:829-32. [ Links ]
20. Percy MJ, Furlow PW, Lucas GS, et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358:162-8. [ Links ]
Endereço para correspondência
Ermelinda Santos Silva
S. Gastrenterologia Pediátrica,
Departamento da Criança e do Adolescente,
Centro Hospitalar do Porto
Largo Prof. Abel Salazar,
4099-001 Porto.
e-mail: ermelinda.dca@chporto.min-saude.pt
telefone: 222 077 500
Recebido a 26.12.2014 | Aceite a 13.02.2015


{kind=link}