










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.22 no.3 Porto set. 2013
QUAL O SEU DIAGNÓSTICO? / WHAT IS YOUR DIAGNOSIS?
Genes, crianças e pediatras
Joana CorreiaI; Marta RiosII; Paula FerreiraII; Esmeralda MartinsIII; Anabela BandeiraIII
IS. Pediatria, CH Porto, 4099-001 Porto, Portugal. jbcorreia@sapo.pt
IIS. Cuidados Intensivos Pediátricos, CH Porto, 4099-001 Porto, Portugal. martariospinho@gmail.com; preginaferreira@gmail.com
IIIU. Doenças Metabólicas, S. Pediatria, CH Porto, 4099-001 Porto, Portugal. eg.esmeralda@gmail.com; anabela.ol.bandeira@sapo.pt
ABSTRACT
A male newborn, presenting hipotonia and posterior parietal bossing, developed, in the first 12 hours of life, refusal to feed and hypoglycaemia. A cranial ultrasound, skull X-ray and CT scan revealed an occipital and parietal fracture with an underlying haematoma and extensive extracranial soft-tissue swelling. He was submitted to surgical drainage. After 24 hours: new intracerebral bleeding. At the age of two-months he presented abnormal skin and sparse kinky hair. Serum copper and caeruloplasmin levels were below the normal range. Molecular diagnosis of Menkes disease was made by the identification of a new mutation in ATP7A gene.
Keywords: Congenital fractures, intracerebral bleeding, sparse kinky hair.
Primeiro filho de um casal jovem não consanguíneo sem história de doenças heredo familiares. Gestação vigiada sem intercorrências até ao 3º trimestre, altura em que foi detetado restrição do crescimento intra uterino.
Parto eutócico hospitalar induzido às 37 semanas. Índice de Apgar 9/9.
Exame objetivo: Peso 2200 g (<P5); Comp. 47 cm (P25-50); P. Cefálico 34 cm (P 75).
Hipotonia axial, bossa parietal posterior. Sem dismorfias e restante exame físico normal. Observado às 12 horas de vida por recusa alimentar e hipoglicemia. Apresentava volumoso cefalohematoma parieto-occipital direito com crepitação occipital.
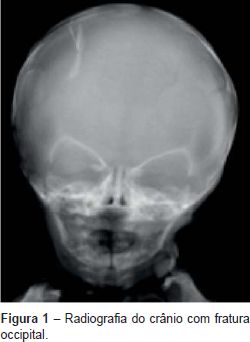
Fez radiografia do crânio (Figura 1) que mostrou fratura occipital.
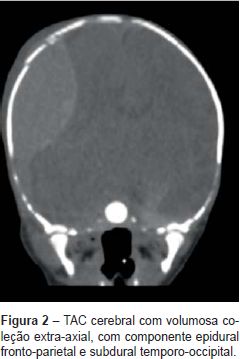
A tomografia axial computorizada (TAC) cerebral (Figura 2) mostrou fratura cominutiva com afundamento occipito-parietal direito; volumosa coleção extra-axial, com componente epidural fronto-parietal e subdural temporo-occipital extenso; deformação do parênquima adjacente e redução dos ventrículos laterais e III ventrículo e foco de contusão hemorrágica parietal esquerdo.
A radiografia toracoabdominal revelou fraturas da clavícula direita, e das costelas. Fez radiografia dos ossos longos que mostrou fratura cubital esquerda e peronial bilateral.
Foi submetido a drenagem do hematoma subdural por Neurocirurgia.
Em D2 apresentava edema marcado da cabeça e pescoço, pelo que repetiu TAC cerebral que revelou novo hematoma subdural agudo retrocerebeloso. Manteve tratamento conservador com evolução clínica e imagiológica favorável. Em D 7 de vida iniciou crises convulsivas medicadas com fenobarbital.
Fez cariótipo de sangue periférico que foi normal (46,XY).
As ecografias renal e abdominal e o ecocardiograma não revelaram alterações.
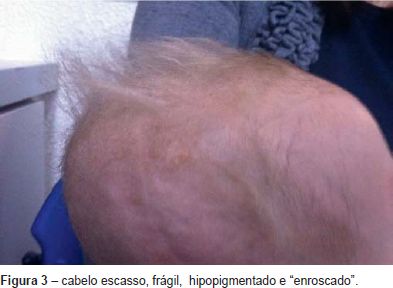
Manteve hipotonia axial marcada, sem controle cefálico com algum contacto social. Pelos dois meses de vida foi notado o aparecimento de um cabelo escasso, frágil, hipopigmentado e enroscado e hipopigmentação cutânea (Figura 3). Não apresentou novas fraturas ósseas.
Qual o seu diagnóstico?
COMENTÁRIOS
Pela presença de fracturas ósseas ao nascimento foi colocada a hipótese de diagnóstico de Osteogénese Imperfeita. No entanto a ausência de história familiar e de escleras azuis, embora não o excluindo, tornam este diagnóstico pouco provável.
Por outro lado, a associação de fraturas ósseas com hemorragia cerebral ao nascimento e a ocorrência de nova hemorragia cerebral fizeram-nos evocar a hipótese de Doença de Menkes, corroborada pela evolução clínica: a criança manteve hipotonia axial marcada com pouco controle cefálico e o aparecimento de um cabelo ralo, hipopigmentado e enroscado e hipopigmentação cutânea (Figura 3).
O doseamento de ceruloplasmina 4mg/dl (N>20mg/dl) e do Cobre 8,44μmol/l (N > 12,5 μmol/L) corroboraram este diagnóstico que foi confirmado por estudo molecular: mutação de novo (c.3868C>T) no gene ATP7A.
A Doença de Menkes é uma doença genética, do metabolismo do cobre, recessiva ligada ao X, causada por mutações no gene transportador de cobre, ATP7A, cuja prevalência varia entre 1 em 100.000 a 250.000 nascimentos.
Caracteriza-se por atingimento degenerativo do Sistema Nervoso Central (SNC) com epilepsia e do tecido conjuntivo com alteração da estrutura do cabelo (fino, esparso e quebradiço) e hipopigmentação cutânea.
Estas características surgem pelos 2-3 meses de idade com posterior atraso do desenvolvimento psicomotor, hipotonia, má evolução ponderal por dificuldades alimentares, hipotermia e dificuldade respiratória.
Pode manifestar-se no período neonatal por fraturas congénitas e cefalohematomas uma vez que as alterações do tecido conjuntivo incluem alterações vasculares, com risco aumentado de hemorragia intracraniana.
Os níveis baixos de ceruloplasmina e cobre apoiam o diagnóstico mas estes achados podem surgir no recém-nascido normal. O estudo molecular estabelece o diagnóstico definitivo.
A reposição com histidina de cobre tem demonstrado alguma eficácia, sendo a resposta dependente da precocidade da sua instituição (primeiro mês de vida) e do genótipo, (melhor se persiste algum transporte residual de cobre).
Estas crianças apresentam uma esperança média de vida inferior a cinco anos.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Tümer Z, Moller LB. Menkes disease. Eur JHum Genet 2010, 18:511-8. [ Links ]
2. Kaler SG, Holmes CS, Goldstein DS, Tang J, Godwin SC, Donsante A, et al. Neonatal diagnosis and treatment of Menkes disease. N Eng J Med 2008: 358;605-14. [ Links ]
Anabela Bandeira
Centro Hospitalar do Porto
Serviço de Pediatria - Unidade de doenças metabólicas
Largo do Prof. Abel Salazar
4099-001 Porto, Portugal
E-mail: anabela.ol.bandeira@sapo.pt
Recebido a 09.10.2013 | Aceite a 25.10.2013

