










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.22 no.2 Porto abr. 2013
CASO CLÍNICO / CASO CLÍNICO
Encefalopatia multiquística – a propósito de dois casos clínicos
Multicystic encephalopathy – considering two case reports
Bárbara PereiraI; Clara Alves PereiraII; Joana PintoII; Manuela SantosIII
IS. Pediatria, H Braga, 4710-243 Braga, Portugal
IIS. Pediatria, CH Tâmega e Sousa, 4564-007 Penafiel, Portugal
IIIS. Neuropediatria, CH Porto, 4099-001 Porto, Portugal
RESUMO
Introdução: A encefalopatia multiquística é uma entidade rara, em que o parênquima cerebral é substituído por quistos de vários tamanhos. Tem sido associada a múltiplas causas: transfusão feto-fetal em gravidez gemelar monozigótica, encefalopatia neonatal, infeções do sistema nervoso central, acidente vascular cerebral, trauma, hipotensão materna severa, entre outras.
O fator comum que liga todas estas situações é a ocorrência de hipóxia cerebral grave. O seu diagnóstico baseia-se em exames de neuroimagem, sendo a ecografia transfontanelar o exame de primeira linha. É uma patologia de mau prognóstico que, na maioria dos casos, evolui para disfunção neurológica severa progressiva até à morte.
Casos clínicos: Os autores apresentam dois casos clínicos de encefalopatia multiquística, ambos de apresentação neonatal precoce mas distinta.
Discussão/Conclusão: O conhecimento desta entidade, dos seus fatores de risco e da sua importante morbimortalidade infantil, são essenciais para o seu reconhecimento precoce e abordagem adequada.
Palavras-chave: Encefalopatia multiquística, epilepsia, lactente.
ABSTRACT
Introduction: Multicystic encephalopathy is a rare disease where the brain parenchyma is replaced by cysts of different sizes. It has been associated to multiple causes: foetal transfusion in monozygotic twin pregnancy, neonatal encephalopathy, central nervous system infections, stroke, trauma, severe maternal hypotension, among others. The common factor linking all of these conditions is the occurrence of severe cerebral hypoxia. The diagnosis is based on neuroimaging and cranial ultrasonography is considered the first-line exam. It is a disease with poor prognosis and in most cases it develops into severe progressive neurological dysfunction to death.
Cases Report: The authors report two cases of multicystic encephalopathy, both with early neonatal presentation but distinct.
Discussion/Conclusion: The knowledge of this entity, its risk factors and its important morbidity and mortality in children are essential for the early diagnosis and appropriate approach.
Keywords: Epilepsy, infant, multicystic encephalopathy.
INTRODUÇÃO
Ao longo da gestação, no momento do parto ou no período pós-natal imediato, o cérebro pode sofrer várias agressões. Os vários tipos de lesões cavitadas correspondem a reações diferentes a esses insultos, e dependem da fase do desenvolvimento do cérebro no momento da sua ocorrência.1 De acordo com a extensão dos danos, podemos classificar as lesões major em porencefalia (lesão cavitada hemisférica, sem estruturas no seu interior), encefalopatia multiquística (uma ou múltiplas cavidades de vários tamanhos envolvendo a maior parte de dos hemisférios cerebrais, revestidas por proliferação astrocitária e com septos gliais) e hidranencefalia (necrose maciça de ambos os hemisférios).1,2
A Encefalopatia multiquística (EM) é uma entidade rara, no entanto, com a evolução dos meios imagiológicos, o seu reconhecimento tem vindo a aumentar.3
A EM tem sido associada a múltiplas causas: transfusão feto-fetal em gravidez gemelar monozigótica (mais frequente se há morte intrauterina de um dos fetos)4,5, encefalopatia neonatal6,7,8 (mais frequentemente encefalopatia hipoxico-isquémica), infeções do sistema nervoso central9,10 (meningite/encefalite e bacteriana/vírica), acidente vascular cerebral4,11, traumatismo12, hipotensão materna severa4, entre outras. O fator comum que liga todas estas situações é a ocorrência de hipóxia cerebral grave13,14.
Clinicamente, a EM pode manifestar-se logo no período neonatal, através de sinais de hiperexcitabilidade inespecífica (choro e irritabilidade) ou mesmo disfunção neurológica grave (crises epiléticas até estado de mal convulsivo e coma)4, ou pode permanecer silenciosa14 e ser apenas diagnosticada mais tarde, quando a criança falha a aquisição dos marcos basilares do desenvolvimento psicomotor14.
O diagnóstico da EM, em vida, é efetuado através de exames de neuroimagem. A ecografia transfontanelar (EcoTF) é o exame de primeira linha4, detetando a presença de quistos, no entanto, a ressonância magnética cerebral (RMc) permite completar a definição estrutural dos achados patológicos4,11. As lesões quísticas localizam-se fundamentalmente na zona de união da substância branca com a substância cinzenta, nos territórios vasculares correspondentes às artérias cerebral anterior e média.4,5 Assim, os gânglios da base, cerebelo e tronco cerebral são geralmente poupados, por estarem dependentes do sistema vertebrobasilar.4
Relativamente ao tratamento da EM, este é sintomático, incluindo cuidados de suporte, numa fase inicial e, posteriormente o controlo da epilepsia, geralmente refratária, a estimulação e a intervenção precoces.
A EM é uma patologia de mau prognóstico13 que, na maioria dos casos, evolui para disfunção neurológica grave progressiva até à morte4.
Os autores apresentam dois casos clínicos de EM, com mecanismos de lesões provavelmente diferentes.
CASOS CLÍNICOS
Caso clínico 1
Recém-nascido (RN) do sexo feminino nascido de uma primeira gestação de termo (38 semanas), vigiada, que decorreu sem alterações serológicas e ecográficas de relevo e sem risco infecioso. O parto foi hospitalar, distócico (ventosa), por bradicardia fetal sustentada. O RN era leve para a idade gestacional (peso 2280 g). O Índice de Apgar foi de 4/5/7 ao 1º, 5º e 10º minuto, respetivamente. Ao nascimento, o RN apresentava-se pálido, hiporreactivo, bradicárdico e estava em apneia. Foi entubado e ventilado, com recuperação da cor e frequência cardíaca. Foi internado na Unidade de Cuidados Intensivos Neonatais (UCIN), já em ventilação espontânea, hemodinamicamente estável e apirético, sendo notória a presença de irritabilidade. Foi objetivada acidose metabólica (GSV - pH 7,01, pCO2 37 mmHg, HCO3-8,9 mmol/L, BE - 23,2 mmol/L,). No segundo dia de vida registou-se dois episódios interpretados como episódios convulsivos (movimentos mastigatórios, pedalagem, abertura dos dedos em leque e hipertonia dos membros), tendo iniciado fenobarbital. A investigação complementar efetuada encontra-se enunciada no Quadro 1. As alterações clínicas, analíticas e imagiológicas permitiram o diagnóstico de encefalopatia hipoxico-isquémica. Não se verificou registo clínico ou eletroencefalográfico de novos episódios convulsivos durante o restante internamento tendo suspendido terapêutica antiepilética ao fim de 28 dias. Teve alta aos 32 dias de vida assintomática. Aos quatro meses reiniciou crises em salva, caracterizadas por versão cefálica e dos olhos preferencialmente para a esquerda, com pestanejo, postura tónica dos membros, geralmente assimétrica com extensão mais marcada à esquerda, com correspondente atividade ictal no eletroencefalograma (EEG). Reiniciou fenobarbital, com controlo parcial das crises. Aos seis meses, altura em que teve a primeira consulta de Neuropediatria, era notória a irritabilidade e o atraso global do desenvolvimento. Não estabelecia contacto ocular e evidenciava hipotonia axial e hipertonia dos membros, com Rossolimo bilateral. Nessa altura repetiu RMc (Quadro 1) que revelou a presença de lesões quísticas cortico-subcorticais fronto-parieto-temporais bilaterais dos núcleos da base e tálamos, o que permitiu o diagnóstico de EM. Manteve crises epiléticas, com necessidade de tratamento com várias associações de fármacos. Atualmente com 14 meses de idade, apresenta atraso do desenvolvimento psicomotor, distonia generalizada grave e epilepsia refratária.
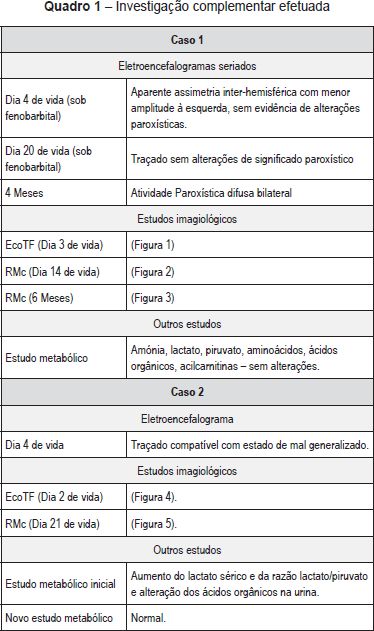
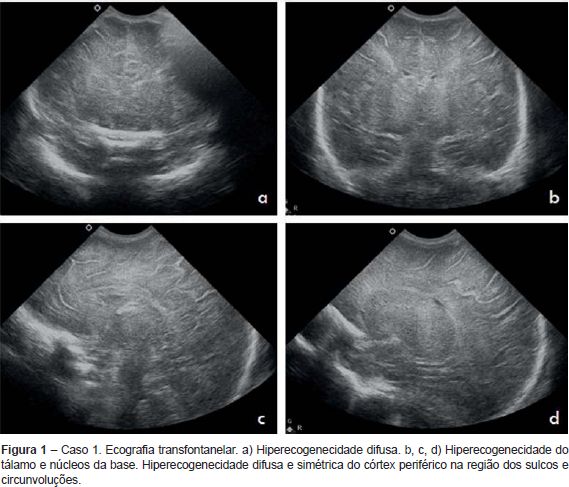
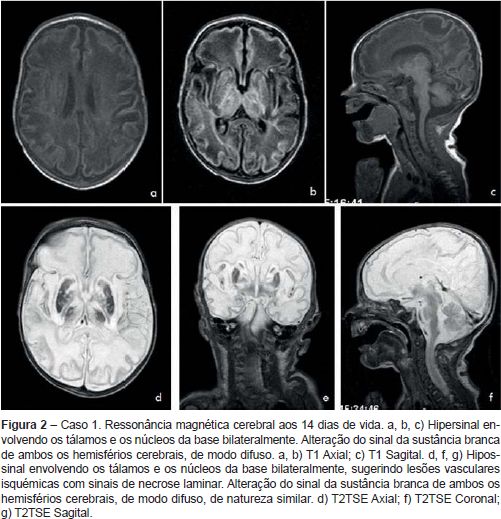
Caso clínico 2
RN do sexo feminino nascido de uma primeira gestação de termo (39 semanas), vigiada, que decorreu sem intercorrências até ao dia anterior ao parto altura em que foi diagnosticado oligoâmnios. Não foram detetadas alterações serológicas nem ecográficas anteriormente. O parto foi hospitalar, distócico (cesariana) por incompatibilidade feto-pélvica e decorreu sem evidência de sinais de sofrimento fetal. A somatometria ao nascimento era adequada à idade gestacional. O índice de Apgar foi 5/8/9 ao 1º, 5º e 10º minutos, respetivamente. Por apresentar depressão respiratória, e ter sido administrada petidina periparto à mãe, fez naloxona com boa resposta clínica permanecendo junto da mãe. Às 14 horas de vida registaram-se dois episódios de cianose, apneia e hipertonia, com duração de cerca de 30 segundos e de resolução espontânea. Foi internado na UCIN para vigilância. Verificou-se a ocorrência de episódios sugestivos de crises epiléticas (pestanejo e nistagmo, e dessaturação), pelo que iniciou fenobarbital. Por manter crises (vários episódios de apneia, cianose, olhar fixo, por vezes com desvio para a direita, hipotonia axial e hipertonia dos membros, com cerca de 20-30 segundos de duração cada), iniciou tratamento com fenitoína. Ao quarto dia realizou EEG, que evidenciou estado de mal elétrico (Quadro 1) tendo-se associado midazolam em perfusão à terapêutica em curso, com resposta. Foi suspensa fenitoína e midazolam uns dias depois. A RMc realizada ao 21º dia revelou múltiplos quistos subcorticais fronto-parietais e na região occipital direita, com perda global do volume encefálico, o que permitiu o diagnóstico de EM (Quadro 1). Não evidenciava lesões a envolver os núcleos da base, os tálamos ou o compartimento infratentorial. Atualmente com nove meses de idade apresenta atraso do desenvolvimento psicomotor, com distonia orolingual, hipertonia dos membros e hiperreflexia osteotendinosa. Mantém tratamento com fenobarbital, tendo controlado as crises.
DISCUSSÃO
A tendência do cérebro imaturo para cavitação depende do tempo e da natureza da agressão. Na EM é mais provável que ocorra entre o terceiro trimestre de gestação e o período pós-natal precoce1,5, depois da migração neuronal, e quando o cérebro já possui a capacidade de produzir uma importante reação glial perante qualquer agressão.4
A EM está associada a uma grande variedade de patologias em que ocorre hipóxia cerebral grave.13,14 O edema resultante agrava a hipoperfusão cerebral e produz perda da capacidade de auto-regulação vascular cerebral, resultando em isquemia segmentar.4 Gravidez gemelar monozigótica com morte intrauterina de um dos fetos, encefalopatia hipoxico-isquémica e infeção do sistema nervoso central são as três causas mais frequentemente associadas a EM, existindo no entanto muitas outras.4,6,9,10
Os autores apresentam dois casos clínicos de EM em recém-nascidos de termo, de gestações únicas e sem intercorrências de relevo. No primeiro caso, a evidência clínica de asfixia neonatal e a distribuição das lesões quísticas, com atingimento dos tálamos e núcleos da base, sugerem a existência de um episódio encefalopático hipóxico-isquémico agudo grave na base etiológica da EM. No segundo caso, a presença de lesões dispersas por ambos os hemisférios cerebrais mas poupando os tálamos e os núcleos da base, bem como a forma de apresentação perinatal apontam para uma agressão de carácter mais arrastado no tempo, que se culminou num estado de mal convulsivo neonatal.
A evolução das técnicas de neuroimagem constituiu um avanço significativo na avaliação do timing da agressão e do reconhecimento do padrão das lesões.8 A RMc permitiu definir dois padrões de lesão em recém-nascidos de termo8: o padrão baso-ganglio-talámico que afeta predominantemente a substância cinzenta profunda cerebral, núcleos da base e córtex sensoriomotor; e o padrão watershed zones que envolve as zonas intravasculares, atingindo a substância branca e a zona parassagital com necrose cortical. O primeiro padrão, também denominado de asfixia aguda quase completa, surge habitualmente associado a um evento sentinela agudo. A asfixia parcial prolongada decorrente de infeções, hipotensão, hipoglicemia, entre outros, e condiciona geralmente o padrão das zonas intravasculares.
Em ambos os casos apresentados, as lesões são imagiologicamente compatíveis com uma agressão do tipo hipoxico-isquémico, no entanto, se no primeiro caso existe evidência clínica da agressão, no segundo não se pode excluir a etiologia metabólica ou infeciosa.
CONCLUSÕES
Os autores descrevem estes dois casos clínicos de EM, pela importante morbimortalidade infantil que esta patologia apresenta, considerando essencial o conhecimento desta entidade e dos seus fatores de risco, de modo a permitir um diagnóstico e intervenção precoces. O estabelecimento do diagnóstico bem como o conhecimento do prognóstico desta patologia vai permitir planear o seguimento do doente e orientar a família. A EM tem mau prognóstico, com agravamento neurológico progressivo até à morte, sendo a mortalidade mais elevada no primeiro ano de vida.4
REFERÊNCIAS BIBLIOGRÁFICAS
1. Low C, Garzon E, Carrete Jr H, Vilanova LC, Yacubian EM, Sakamoto AC. Early destructive lesion in the developing brain. Clinical and electrographic correlates. Arq Neuropsiquiatr 2007; 65:416-22. [ Links ]
2. Aicardi J. Disease of the nervous system in childhood. 1st ed. London/Oxford/New York: Mac Keith Press, 1992: 21-2. [ Links ]
3. Frigieri G, Guidi B, Zaccarelli SC, Rossi C, Muratori G, Ferrari F, et al. Multicystic encephalomalacia in term infants. Childs Nerv Syst 1996; 12:759-64. [ Links ]
4. Orejón G, Beato FM, Heras RS, Molina MM. Encefalomacia multiquística. Revisión de 19 casos. An Esp Pediat 1997; 46:33-9. [ Links ]
5. Gathwala G, Magu S, Khosla A. Multicystic Encephalomalacia. Indian J Pediatr 2003; 70:443-5. [ Links ]
6. Cowan F, Rutherford M, Groenendaal F, Eken P, Mercuri E, Bydder GM, et al. Origin and timing of brain lesions in term infants with neonatal encephalopathy. The Lancet 2003; 361:736-42. Disponível em: www.thelancet.com/journals/lancet/article/PIIS0140-6736(03)12658-X/fulltext [ Links ]
7. García-Alix A, Biarge MM, Arnaez J, Valverde E, Quero J. Asfixia intraparto y encepalopatía hipóxico-isquémica. Protocolos Diagnóstico Terapéuticos de la Asociación Española de Pediatria: Neonatología. Protocolos actualizados al año 2008. Disponível em: www.aeped.es/sites/default/files/documentos/26.pdf [ Links ]
8. Vries LS, Jongmans MJ. Long-term outcome after neonatal hypoxic-ischaemic encephalopathy. Arch Dis Child Fetal Neonatal Ed 2010;95:F220-4. [ Links ]
9. Chang Y, Soffer D, Horoupian DS, Weiss LM. Evolution of post-natal Herpes simplex virus encephalitis to multicystic encephalopathy. Acta Neuropathol 1990; 80:666-70. [ Links ]
10. Gray PH, Tudehope DI, Masel J. Cystic encephalomalacia and intrauterine herpes simplex virus infection. Pediatr Radiol 1992; 22:529-32. [ Links ]
11. Prager A, Roychowdhury S. Magnetic resonance imaging of the neonatal brain. Indian J Pediatr 2007; 74:173-84. [ Links ]
12. Matlung SE, Bilo RA, Kubat B, van Rijn RR. Multicystic encephalomalacia as an end-stage finding in abusive head trauma. Forensic Sci Med Pathol 2011; doi:10.1007/s12024-011-9236-7. [ Links ]
13. Gupta RK, Pant CS, Sharma A, Khalilullah A. Ultrasound diagnosis of multiple cystic encephalomalacia. Pediatr Radiol 1988; 18:6-8. [ Links ]
14. Stannard MW, Jimenez JF. Sonographic recognition of multiple cystic encephalomalacia. AJR Am J Roentgenol 1983; 141:1321-4. [ Links ]
Bárbara Pereira
Hospital de Braga
Serviço de Pediatria
Sete Fontes – São Victor
4710-243 Braga, Portugal
E-mail: bsgopereira@gmail.com
Recebido a 06/02/2012 | Aceite a 16/08/2012

