










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754
Nascer e Crescer vol.21 no.3 Porto set. 2012
Pulmão e doenças sistémicas: Vasculites
M.Guilhermina Reis1
1S. Pediatria, CH Porto
As manifestações pulmonares ocorrem em quase todas as doenças reumáticas da infância, seja secundariamente à doença subjacente, seja como efeito adverso do tratamento. Os sintomas na criança podem ser subtis e só surgirem com o tempo e, embora pouco frequentes, são uma causa importante de morbimortalidade nestes doentes. Requerem um elevado índice de suspeição e antecipação diagnóstica.
O sistema respiratório pode estar envolvido em todas as vasculites sistémicas, embora com frequência variável. As vasculites pulmonares referem-se à inflamação de vasos de qualquer diâmetro, enquanto as capilarites pulmonares apenas se referem à microcirculação pulmonar. Ambas podem ocorrer nas vasculites sistémicas e nas doenças reumáticas. Mais frequentemente as vasculites pulmonares ocorrem nas vasculites sistémicas que afectam predominantemente pequenos vasos, que se manifestam sobretudo por alterações pulmonares e/ou renais.
A hemorragia alveolar difusa (HAD) associada a vasculite é um evento, potencialmente ameaçador de vida que após episódios repetidos poderá levar a fibrose pulmonar.
Serão abordados nesta apresentação:
1. Identificação de sinais e sintomas sugestivos de vasculite pulmonar;
2. Avaliação clínica e laboratorial nas vasculites pulmonares;
3. Princípios do tratamento nas vasculites pulmonares.
As vasculites sistémicas são raras, com incidências de 20-100 casos/milhão e prevalência de 150-450 casos/milhão. A mais recente classificação das vasculites na criança resulta dos critérios de Consenso da EULAR e Sociedade Europeia de Reumatologia Pediátrica (Quadro I).
Quadro I – Nova classificação de vasculites em idade pediátrica*

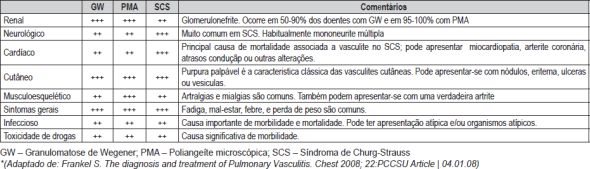
A doença pulmonar é muito comum e uma característica importante nas vasculites associadas à presença de Anticorpos anti-citoplasma dos neutrófilos (vasculites ANCA+), tais com a Granulomatose de Wegener (GW), Síndrome de Churg-Strauss (SCS) e Poliangeíte microscópica (PM).
Nas vasculites de grandes vasos o pulmão raramente é afectado: na arterite de células gigantes a manifestação respiratória mais frequente é a tosse, geralmente não produtiva, persistente e que responde aos corticosteroides; na Arterite de Takayasu, o envolvimento respiratório é frequentemente subclínico e detectável por técnicas não invasivas.
O envolvimento pulmonar é raro na Poliarterite nodosa, Doença de Kawasaki, Purpura de Henoch-Schönlein (PHS) e na vasculite crioglobulinemica.
As vasculites pulmonares acompanham-se frequentemente de manifestações sistémicas, incluindo astenia, febre, perda ponderal, artralgias, manifestações renais e exantemas. A hemorragia alveolar difusa (HAD) associada a vasculite, por lesão da microcirculação alveolar (infiltração vascular neutrofilica dos septos peribronquiolares e interalveolar, com a activação do processo inflamatório; disrupção anatómica capilar e libertação de enzimas proteoliticas...) com formação de fibrina e necrose fibrinoide do interstício, é um evento, potencialmente ameaçador de vida que, após episódios repetidos, poderá levar a fibrose pulmonar. Pode ser maciça e manifesta-se habitualmente por anemia, hemoptises, infiltados alveolares na radiografia torácica e, se grave, por insuficiência respiratória hipoxémica. A glomerulonefrite pode também ser rara com prodromos frustes. (Quadro II)
Quadro II – Manifestações sistémicas das vasculites*
Qualquer criança com anemia e infiltados pulmonares deve ser suspeita de ter hemorragia alveolar. Na pele devem procurar-se sinais de vasculite: purpura palpável, eritema nodoso, ulcerações, hemorragias subungueais; no nariz, orelhas e naso/orofaringe sinais de hemorragia ou granulomas. No sedimento urinário avaliar a presença de proteinúria, hematúria e cilindros.
O estudo imunológico com pesquisa de anticorpos (Ac´s) é fundamental no diagnóstico das vasculites de pequenos vasos. O diagnóstico de Sindrome de Goodpasture (SG) assenta na presença de Ac´s anti membrana basal glomerular; enquanto a GW, PM e outras vasculites se associam à presença de anticorpos anti-citoplasma de neutrófilos (ANCA). A determinação da ANCA por Imunofluorescência indirecta (IIF) deve ser complementada por técnicas de ELISA (Enzyme-Linked Immunosorbent Assay - Ensaio de Imunoabsorção Ligado a Enzima), já que a positividade por IIF é inespecífica, podendo ocorrer noutras patologias: Lupus eritematoso sistémico (LES), Artrite Idiopática Juvenil (AIJ), etc. O teste de ELISA permite a diferenciação de c-ANCA ou PR3-ANCA e p-ANCA ou MPO-ANCA. Os ANA e Factor Reumatoide (FR) podem ser positivos nas vasculites primárias, no entanto títulos elevados associados à presença de Ac´s específicos (dsDNA, Anti-RNP, anti-JO-1,etc) podem identificar uma doença reumática subjacente. A determinação da IgE é importante quando se coloca a hipótese de CSS.
A radiografia torácica ou TC (tomografia computorizada) podem evidenciar alterações ainda antes de manifestações clínicas significativas; estas alterações são geralmente inespecíficas. Na suspeita de HAD a identificação de cavidades, nódulos ou padrão difuso em vidro despolido deve colocar a suspeita de vasculite.
A broncofibroscopia deve ser ponderada quando a clínica e achados radiológicos, deixam dúvidas entre hemorragia versus infecção.
A realização de biópsia pulmonar raramente será necessária na presença de outras alterações sistémicas, já que o risco será menor se efectuada biópsia cutânea ou renal. Já nas doenças auto-imunes sistémicas a doença pulmonar crónica pode ser a primeira manifestação e assim os achados histológicos poderão ser importantes para confirmar achados de vasculite ou procurar outras causas de HAD.
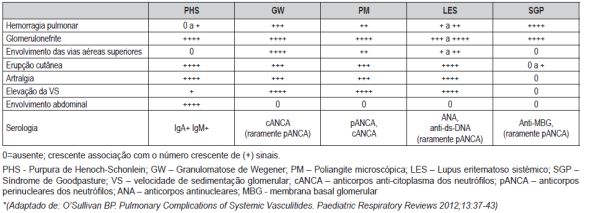
Os síndromes pulmão-rim resultam das vasculites de pequenos vasos, estando habitualmente associados a componente inflamatório. São raros, mas exigem um elevado índice de suspeição, já que o seu reconhecimento permite instituir tratamento anti-inflamatório que é life-saving. (Quadro III)
Quadro III - Achados clínicos e serológicos em doentes com síndromes de pulmão-rim*
A Purpura de Henoch-Schonlein (PHS) é a mais frequente vasculite de pequenos vasos (incidência de 10-14/100000) e a que menos se associa a manifestações pulmonares; apresenta-se com pápulas eritematosas, seguida de púrpura palpável, não trombocitopenica nos membros inferiores, tronco e face. Há envolvimento renal em 60% casos. (Quadro IV)
Quadro IV – Critérios de classificação de Purpura Henoch-Schönlein*

A hemorragia pulmonar é secundária à disrupção alveolo-capilar pelos complexos imunes circulantes. A HAD e a pneumonite intersticial são raras; Após o diagnóstico de PHS alguns doentes tem manifestações pulmonares ligeiras como tosse, que pode ser hemoptoica ou crepitações na auscultação pulmonar, mas pode haver HAD suficiente para necessitar de Ventilação mecânica e transfusão de hemáceas. O tratamento pode requerer, para além de corticoterapia (Cct), a utilização de ciclofosfamida, nos casos de hemorragia grave.
A hemorragia alveolar difusa (HAD) pode ocorrer na Purpura de Henoch Schonlein (PHS), no Sindrome de Goodpasture (SG), vasculite crioglubinémica (VC) e outras colagenoses.
A oclusão da artéria pulmonar devido a trombose e aneurismas arteriais pulmonares são complicações graves da Doença de Beçhet (DB).
A Granulomatose de Wegener (GW) caracteriza-se pela tríade de inflamação granulomatosa das vias aéreas altas e baixas, vasculite necrotizante e PR3-ANCA; é rara na criança, que frequentemente apresenta febre, artralgias ou artrite e exantemas cutâneos ou úlceras. As manifestações cutâneas podem preceder em meses outras manifestações.
O atingimento respiratório é amplo: rinorreia (purulenta/sanguinolenta), secura nasal, epistaxis, sinusite, otite, nariz em sela por perfuração do septo nasal; tosse, hemoptise, dispneia, dor torácica (pleurítica); estridor por estenose subglótica ou traqueobrônquica pode ser o sinal de apresentação. A hemorragia alveolar é rara, mas pode ser maciça; o atingimento renal quando surge requer tratamento agressivo para evitar evolução para insuficiência renal terminal.
A doença pulmonar desenvolve-se em cerca de 75% das crianças com GW; um terço das alterações radiológicas nas crianças com GW, não se associa a sintomas pulmonares; nódulos e massas pulmonares, solitários ou múltiplos, são os achados mais frequentes na radiografia e TAC torácicos. (Quadro V)
Quadro V – Critérios de classificação para Granulomatose de Wegener*

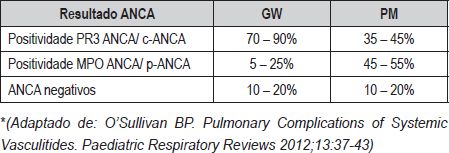
Os testes laboratoriais são inespecíficos na GW, excepto PR3-ANCA. Se clínica sugestiva, o PR3-ANCA tem valor preditivo positivo de 99% e pode ser dispensável o diagnóstico histológico para instituir tratamento.
A Poliangeite microscópica (PM) é uma vasculite necrotizante dos pequenos vasos, não granulomatosa e sempre associada a glomerulonefrite segmentar e focal. A sua incidência é de 1:100.00 e há escassos casos relatados na Pediatria. A hemorragia alveolar difusa (HAD), expressa por hemoptises, dificuldade respiratória e anemia, devida à capilarite alveolar, ocorre em 1/3 dos doentes e é a manifestação mais frequente do envolvimento pulmonar. A HAD pode ser subclinical, devendo suspeitar-se na presença de infiltrados alveolares bilaterais de novo, na radiografia torácica, associados à descida no valor de hemoglobina. Verificam-se grandes atrasos no diagnóstico pela inespecificidade dos sintomas: febre, perda ponderal, mialgias, artralgias; a hemorragia alveolar pode surgir isoladamente; a fibrose pulmonar intersticial pode preceder em anos a vasculite pulmonar e ser a primeira manifestação de PM.
O seu diagnóstico serológico é mais difícil do que na GW. (Quadro VI)
Quadro VI – Distribuição da Serologia ANCA na GW e na PM*
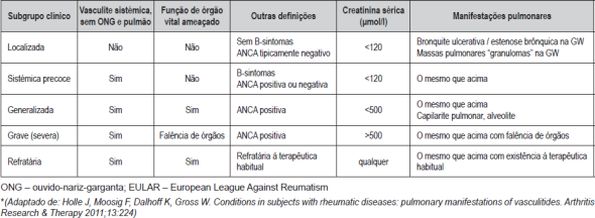
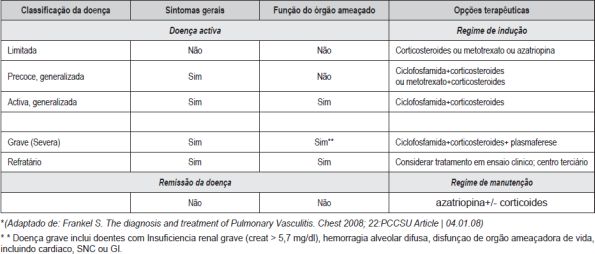
O tratamento das vasculites ANCA+ (GW e MPA), desconhecida a sua etiologia, visa actualmente, a indução da remissão. As estratégias terapêuticas dependendo do estadio clínico. (Quadro VII)
Quadro VII – Definição dos estadios da doença nas vasculites ANCA associadas, de acordo com o EULAR*
A corticoterapia (Cct) sistémica e a ciclofosfamida são a base terapêutica para a indução da remissão na doença sistémica (tal como na capilarite pulmonar), podendo ter de se fazer recurso da plasmaferese ou de Ac´s monoclonais (Rituximab) nas vasculites refractárias. Há evidência para uso de terapêutica de manutenção (Metotrexato, Azatioprina, Micofenolato de Mofetil) em conjunto com a Cct na GW e na PM. O advento das terapias biológicas traz esperança para melhor controle destas entidades. As infecções respiratórias continuam a ser um problema major nos doentes com vasculites sob imunossupressão intensiva, incluindo uso de agentes biológicos. A profilaxia da infecção por Pneumocistis jirovecci parece ser eficaz nos doentes sob ciclofosfamida. (Quadro VIII)
Quadro VIII – EUVAS – Classificação da gravidade das doenças e opções de tratamento*
O Síndrome de Churg-Strauss (SCS) ou angeíte granulomatosa alérgica é um síndrome pauciimune pulmão-rim. Define-se por doença granulomatosa eosinofílica ANCA-associada, que envolve o pulmão e seios perinasais. O sintoma cardinal é a asma, frequentemente precedida de rinite alérgica, frequentemente complicada de polipose nasal e sinusite.
A tríade infiltração tecidular eosinofilica, vasculite necrotizante e granulomas extravasculares, raramente ocorre em simultâneo.
O SCS é raro, sendo desconhecida a sua incidência e prevalência em Pediatria.
Para o diagnóstico requerem-se quatro dos seis critérios (sensibilidade 85%, especificidade>99%). (Quadro IX)
Quadro IX – Critérios de classificação do Síndrome de Churg-Strauss*

A apresentação clínica do CSS ocorre por fases, que podem distanciar-se em muitos anos: 1ª fase ou prodrómica com rinite e asma, 2ª fase eosinofilia periférica e tecidular e 3ª fase com vasculite sistémica. Na fase vasculítica pode ocorrer miocardite e pericardite, verificando-se tamponamento cardiaco em cerca de metade dos doentes. As manifestações cutâneas podem atingir 75% doentes (purpura, nódulos subcutâneos). O atingimento renal é menos grave e menos frequente na WG.
Infiltrados pulmonares alveolares focais e transitórios, são o achado radiográfico mais comum, sendo menos frequente a HAD. Na 2º fase da doença pode haver padrão de pneumonite eosinofilica crónica.
Os exames laboratoriais evidenciam eosinofilia e elevação da velocidade de sedimentação, podendo ser MPO-ANCA positivo.
As crianças são mais frequentemente ANCA negativo. Pode ser necessária a biópsia pulmonar se o pulmão é o único órgão envolvido, sendo que a biópsia transbronquica pode não ser produtiva, tendo de equacionar-se biopsia por video-toracoscopia.
O tratamento assenta na Cct, podendo ser considerados outros imunossupressores nos casos refractários.
As vasculites pulmonares são raras e condições ameaçadores de vida. Se reconhecidas, podem ser tratadas com sucesso.
BIBLIOGRAFIA
1. Robin Deterding et al. Rheumatic and Granulomatous Diseases. Pediatric Pulmonology, American Academy of Pediatrics, 2011 [ Links ]
2. O Sullivan BP. Pulmonary complications of Systemic vasculitides. Paediatr Respir Rev 2012. doi: 10: 1016/j.prrv.2011.04.002 [ Links ]
3. Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K, Davin JC, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis 2006;65:936-41. [ Links ]
4. Manganelli P, Fietta P, Carotti M, Pesci A, Salaffi F. Respiratory system involvement in systemic vasculitides. Clin Exp Rheumatol 2006; 24:S48-59. [ Links ]
5. Kevin K Brown. Pulmonary vasculitis. Proc Am Thorac Soc 2006;3:48-57. [ Links ]
6. Holle JU, Moosig F, Dalhoff K, Gross WL. Conditions in subjects with rheumatic diseases: pulmonary manifestations of vasculitides. Arthritis Research & Therapy 2011;13:224. [ Links ]
7. Demirkaya E, Luqmani R, Ayaz NA, Karaoglu A, Ozen S, et al. Time to focus on outcome assessment tools for childhood vasculitis. Pediatric Rheumatol Online J 2011; 9:29. [ Links ]
8. Frankel S. The diagnosis and treatment of Pulmonary Vasculitis. Chest 2008; 22:PCCSU Article | 04.01.08 [ Links ]
9. Rabinovich CE. Pulmonary complications of childhood rheumatic disease. Paediatr Respir Rev 2012; 13:29-36. [ Links ]
10. Esposito S, Corona F, Defilippi A, Petaccia A, Chidini G, DellEra L, et al. Wegener´s granulomatosis presenting with life-threatning lung hemorrhage in a 7-year-old child. Rheumatol Int 2010; 30: 1665-8. [ Links ]
11. Jindal G, Cruz SD, Punia RP, Kaur R. Refractory anemia as a presenting feature of microscopic polyangiitis: a rare vasculitis in children. Indian J Pediatr 2011; 78:1287-9. [ Links ]

