








Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.34 no.3 Lisboa Sept. 2020
https://doi.org/10.32932/pjnh.2020.10.089
CASE REPORT
Daratumumab: A new future for AL-Amyloidosis and Multiple Myeloma
Clara Pardinhas1, Luís Francisco2, Luís Escada1, Vítor Sousa3, Rui Alves1
1 Nephrology Department, Coimbra University Hospital Center. Faculty of Medicine of the University of Coimbra
2 Hematology Department, Coimbra University Hospital Center
3 Pathology and Cell Biology Department, Coimbra University Hospital Center. Faculty of Medicine of the University of Coimbra
ABSTRACT
Concomitant development of symptomatic amyloid light-chain (AL) amyloidosis and multiple myeloma (MM) is rare and has a poor survival rate. Introduction of thalidomide and bortezomib, a proteasome inhibitor, has improved the survival of patients with MM; however, it has not cured the disease. Nevertheless, monoclonal antibodies may treat the disease using an immunotherapeutic approach. This report describes the case of a 39-year-old man with MM IgG lambda, heavily treated with multiple QT cycles and two hematopoietic stem cell transplants, who developed light chain amyloidosis. After an interdisciplinary discussion, daratumumab was initiated. Approximately 24 months later, he presents complete remission of the disease, normal renal function and no associated symptoms.
Keywords: Light-chain amyloidosis, Multiple Myeloma, Daratumumab, Prognosis
INTRODUCTION
Both multiple myeloma (MM) and immunoglobulin light chain (AL) amyloidosis are plasma cell proliferative disorders that rarely coexist. Approximately 10-15% of patients with MM develop clinical evident AL amyloidosis, with the reverse situation very rare (0.4%).1
Immunoglobulin light chain amyloidosis (AL) results from extracellular deposition of light chain-composed amyloid fibrils leading to organ dysfunction, with significant associated morbidity and mortality, especially for those patients with relapsed or refractory disease with cardiac involvement.2,3 Despite therapeutic advances, AL amyloidosis remains a challenging disease to treat and newer therapies are needed.2
Light chains produced from plasma cells can become amyloidogenic among some patients with MM, thereby leading to systemic amyloidosis.4 In a patient with multiple myeloma, the presence of nephrotic syndrome, congestive myocardial disease, peripheral (including carpal tunnel) or autonomic neuropathy, hepatomegaly, or characteristic findings of macroglossia and periorbital ecchymoses should lead to suspicion of AL amyloidosis.1 In a recent study, in 4,319 MM patients followed for at least six months from 1990 to 2008, only 1.1% (n = 47) had AL amyloidosis.1 Although AL amyloidosis is not a myeloma-defining event (MDE), outcomes of patients with AL amyloidosis who have clonal bone marrow plasmacytosis ≥10 percent are comparable to patients meeting criteria for coexisting MM due to the presence of MDEs.5 Therefore, such patients with AL amyloidosis without clinical myeloma who have a higher plasma cell burden (≥10 percent) should be managed more intensively than their low tumor burden counterparts. Although typical myeloma regimens are often not well tolerated in patients with AL amyloidosis, induction and maintenance strategies should be considered for those who meet the current criteria for coexistent myeloma.5 The coexistence of MM and AL amyloidosis is a poor prognostic factor and there are series that point to an average survival of 1.1 years vs. 2.9 years in cases of MM without amyloidosis. Other studies have shown similar 2.5-year survivals.1
The introduction of the monoclonal antibodies seem to change the paradigm of MM treatment, highlighting the possibility to cure patients using an immunotherapeutic approach.6 The addition of daratumumab to lenalidomide and dexamethasone significantly lengthened progression-free survival among patients with relapsed or refractory multiple myeloma.7 Daratumumab, both as monotherapy and in combination, appears safe and highly effective in heavily pretreated AL amyloidosis. Rapid achievement of hematologic response (HR) with daratumumab-based therapy, particularly with combination therapy, is key.2 Induction therapy with daratumumab in transplant ineligible patients may also improve patient status to allow consideration for hematopoietic stem cells transplantation (ASCT).8 In addition, the favorable toxicity profile makes it a particularly valuable agent for AL amyloidosis with major organ dysfunction, such as cardiac and/or renal.2 However, more studies are needed to verify duration of hematologic responses, organ response and long-term safety and tolerability.
Here, we describe the case of a 39-year-old man with MM IgG lambda, heavily treated with multiple QT cycles and two hematopoietic stem cell transplants, that developed light chain amyloidosis. Daratumumab was initiated and approximately 24 months later, he presents complete remission of the disease, which is a much longer survival than previously reported.1
CASE REPORT
A 39-year-old male patient with clinical history of multiple myeloma immunoglobulin (Ig)G lambda, diagnosed in 2010, underwent multiple chemotherapy regimen cycles with vincristine, carmustine, melphalan, cyclophosphamide and prednisone alternating with vincristine, carmustine, doxorubicin, dexamethasone (VBMCP/VBAD) and two hematopoietic stem cell transplantations (SCT) in 2012. No other known conditions. Evidence of disease progression since March 2017. Protein electrophoresis found hypogammaglobulinemia, increased monoclonal protein with 23.4g/L of serum IgG and a reduced kappa/lambda relation (0.12). Bone marrow aspirate showed 35% clonal plasma cells. In June 2017 he had a pathological fracture of the proximal diaphysis of the left humerus requiring admission at ortho-oncology ward. He was then proposed for lenalidomide and dexamethasone by his hematologist and was waiting for daratumumab approval. The patient was admitted in the emergency room in September 2017 for new onset nephrotic syndrome.
He presented with exuberant peripheral edema, but remaining physical examination was unremarkable. Laboratory studies on admission showed normal renal function (creatinine 0.7mg/dl BUN 40mg/dl), hemoglobin level 13.6 g/dL, white blood cells count 5200/μL with normal differential leukocyte count, total protein 3.9 g/dl, and albumin concentration 1.7 g/dl. He also had total cholesterol of 668mg/dl and triglycerides of 222mg/dl. The dip stick urine examination revealed 3+ protein. A renal ultrasound showed normal sized kidneys without hydronephrosis. He was admitted to Nephrology Department for study of new onset nephrotic syndrome.
Subsequent tests showed a protein/creatinine ratio of 24g/g, presence of a serum monoclonal protein, and an abnormal serum free light chain ratio (kappa/lambda 0.14). Autoimmunity study, serologies and virology tests were all negative. A renal biopsy was performed and showed 8 glomeruli, with amorphous and eosinophilic material infiltration and matrix expansion, sketching nodular structures.
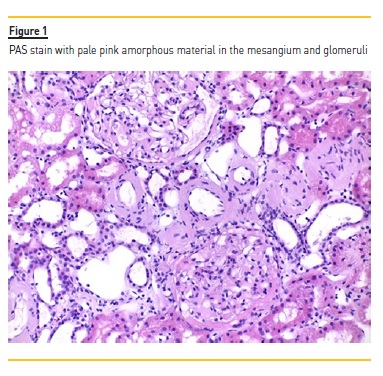
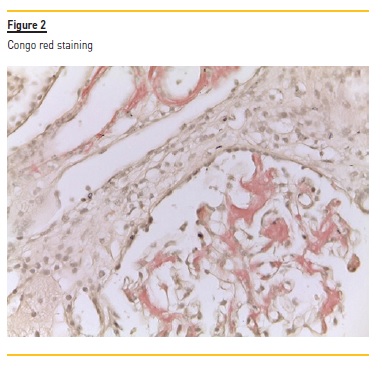
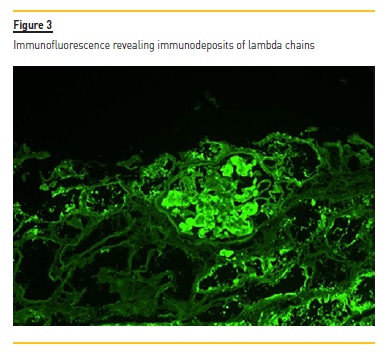
No hypercellularity and no visualization of fibrosis by the Masson’s trichrome technique was observed. There were also signs of tubular atrophy, and vessel wall thickening which was occupied by the same material. The PAS showed pale pink amorphous material in the mesangium. These deposits showed Congo red positivity with apple-green birefringence under polarized light, suggestive of the presence of amyloid substance. Immunofluorescence showed low dense focal deposits of C3 and C1q and there were immunodeposits of lambda chains. According to these findings, we diagnosed the patient with AL amyloidosis associated with multiple myeloma. An echocardiogram was performed and showed no ventricular wall thickening and no granular sparkling; also BNP/pro-BNP levels were normal.
The development of AL amyloidosis was an unexpected complication of primary MM. As this was a rare case in a young patient, we decided that an interdisciplinary discussion would be the best way to proceed. Previous therapy with lenalidomide and dexamethasone was continued, and daratumumab was initiated. Follow-up was maintained after discharge and we observed renal function preservation (serum creatinine 0.9mg/dl) and progressive improvement of hypoalbuminemia and proteinuria.
Two years later, the patient had maintained a biological response with a decrease in the serum lambda chain value. Proteinuria/creatinine ratio improved to 0.38 g/g, and albumin increased to 4.6 g/dL.
He is now waiting for hematopoietic stem cells transplantation (ASCT).
DISCUSSION
Despite being the most common type of systemic amyloidosis in developed countries, AL amyloidosis has an estimated incidence of 9 cases/million inhabitant/year.9 The diagnosis of symptomatic AL amyloidosis in MM patients reaches 10 to 15%. In a recently published study, the incidence is much lower (1.1%).1 Although coexistence is infrequent, it has a very poor survival rate. Despite therapeutic advances in the last decades, these remain two challenging diseases to treat.1
MM is a hematological malignancy characterized by a high tendency to relapse and to become drug resistant. In the past, melphalan was considered the standard treatment for MM patients.6 The introduction of thalidomide and the proteasome inhibitor bortezomib led to a significant improvement of the survival of MM patients: however, none of them reached the cure of the disease. These drugs introduced the concept of targeting not only the malignant clone but also the microenvironment. In addition, the introduction of lenalidomide expanded this concept by focusing on the immune microenvironment.
More recently, the introduction of the monoclonal antibodies seemed to change the paradigm of MM treatment, highlighting the possibility of curing MM patients by an immunotherapeutic approach.6
The addition of daratumumab to lenalidomide and dexamethasone significantly lengthened progression-free survival among patients with relapsed or refractory MM.7 In a recent study, the Kaplan-Meier rate of progression-free survival at 12 months was 83.2% (95% CI, 78.3 to 87.2) in the daratumumab group, as compared with 60.1% (95% CI, 54.0 to 65.7) in the control group. A significantly higher rate of overall response was observed in the daratumumab group than in the control group (92.9% vs. 76.4%, P<0.001), as was a higher rate of complete response (43.1% vs. 19.2%, P<0.001).7 It has an established role as monotherapy with an overall response-rate (ORR) of 30% in heavilypretreated MM patients. In combination with alkylating-agents, proteasome-inhibitors and immunomodulatory-agents, daratumumab is a first-in-class, human IgG1ĸ monoclonal antibody that targets the CD38 epitope.2 It was approved by the U.S. FDA in 2015 and daratumumab is currently approved in combination with dexamethasone plus either lenalidomide, bortezomib or pomalidomide for the treatment of relapsed MM patients.10 Daratumumab has a broadspectrum killing activity in MM, engaging complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and apoptosis.
Moreover, daratumumab modulates the enzymatic activity of CD38 and induces an immunomodulatory role in MM by depleting CD38+ immune suppressive cells contributing to its antitumor activity.10 The CD38 molecule is an ectoenzyme that generates immunosuppressive adenosine which is inhibited by daratumumab, thus improving disease control.10 Daratumumab, both as monotherapy and in combination, appears safe and highly effective in heavily pretreated AL amyloidosis. Rapid achievement of hematologic response with daratumumab-based therapy is significant.11
Notably, these results appear to be far superior to those observed in MM, highlighting the value of targeting CD38 in AL amyloidosis, a disease with substantially lower level of clonal plasmacytosis. In addition, the favorable toxicity profile makes it a particularly valuable agent for AL associated with major organ dysfunction.2
The need to achieve deeper responses to completely turn off production of the amyloidogenic light chain in AL amyloidosis makes daratumumab an attractive option to consider.8 In recent years, several retrospective reports or isolated cases have demonstrated efficacy and tolerability of daratumumab in heavily treated AL amyloidosis patients.3 Several reports have confirmed its efficacy and prospective data are now starting to emerge regarding the safety and efficacy of daratumumab in AL amyloidosis.8 Sanchorawala et al. presented preliminary results of their phase II study in a cohort of 12 patients with relapsed AL amyloidosis, prospectively enrolled to receive daratumumab.
They report rapid reduction in serum free light-chain levels with 10 of 12 patients achieving levels consistent with a very good partial response (VGPR) after a single infusion of daratumumab. Also, a multicenter phase II study across France and Italy reports an overall response rate of 59% with 44% of patients achieving at least a VGPR after at least one cycle of therapy. Kimmich et al. reported the efficacy of daratumumab monotherapy in 48 previously treated (median 3 prior lines of therapy) patients with AL amyloidosis, with 65% patients achieving a hematologic response.8
Recently, the case of two patients with histologically confirmed AL amyloidosis and advanced cardiac involvement treated with daratumumab monotherapy was reported. Both tolerated the therapy without serious adverse events and achieved normal light-chain levels within one cycle of therapy.12 Also, another recent case report showed an NT- pro BNP response in a rapid and effective manner and a rapid complete hematologic response with daratumumab-pomalidomide-dexamethasone.13
In addition, induction therapy with this regimen in transplantineligible patients has been shown to improve patient status to allow consideration for ASCT8, as in the case reported here. The phase 3 ANDROMEDA study is now evaluating daratumumab- cyclophosphamide, bortezomib, and dexamethasone (CyBorD) vs CyBorD in newly diagnosed AL amyloidosis, and preliminary efficacy was robust with high rates of deep and durable hematologic responses by daratumumab plus CyBorD.14
In summary, we herein presented an unusual case of coexistence of both multiple myeloma and light chain amyloidosis, in which daratumumab induced not only a rapid and deep complete response but also a durable one, significantly more prolonged with respect to the previous approaches. Our case suggests that although there is a big diversity and complexity of light chain amyloidosis and multiple myeloma pathophysiology and coexistence, and although the prognosis is poor, small steps are being taken to improve clinical outcomes. The efficacy and tolerability of daratumumab for the treatment of myeloma has led to rapid implementation of this new drug for the treatment of relapsed-refractory MM and AL amyloidosis.9 Daratumumab has shown itself to outperform other novel agents in achieving rapid and deep responses in both AL amyloidosis and MM, and in its potential for treatment of patients that traditionally have a poor prognosis.8
We envision it being increasingly used earlier in the disease course over the coming years. Ultimately, more prospective data are required to demonstrate its superiority as a standard of care in AL amyloidosis.
References
1. Alcatrão MJ, Neves C, Gaspar A, Bravo A, Margarido E, Estrada H. Multiple Myeloma and AL Amyloidosis. Rev da Soc Port Med interna. 2016;23(1):28–31. [ Links ]
2. Abeykoon JP, Zanwar S, Dispenzieri A, Gertz MA, Leung N, Kourelis T, et al. Daratumumab-based therapy in patients with heavily-pretreated AL amyloidosis. Leukemia [Internet]. 2019;33(2):531–536. Available from: http://dx.doi.org/10.1038/s41375-018-0262-2 [ Links ]
3. Canichella M, Serrao A, Annechini G, D’Elia GM, De Luca ML, Pulsoni A. Long-term response to daratumumab in a patient with advanced immunoglobulin light-chain (AL) amyloidosis with organ damage. Ann Hematol. 2019;98(4):1047–1048.
4. Kato H, Fujigaki Y, Asakawa S, Yamaguchi Y, Uozaki H, Komatsuda A, et al. Rapid deterioration of the renal function caused by the coexistence of intratubular amyloidosis and myeloma cast nephropathy. Intern Med. 2015;54(23):3023–3028. [ Links ]
5. Rajkumar SV. UpToDate: Clinical presentation, laboratory manifestations, and diagnosis of immunoglobulin light chain (AL) amyloidosis (primary amyloidosis). UpToDate. 2016;1–21. [ Links ]
6. Past HT, Contributions A. in Immunology (https://www.frontiersin.org/articles/10.3389/fimmu.2019.01945/full. Editorial: Immunotherapy in Multiple Myeloma. 2019;1–8. [ Links ]
7. Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319–1331. [ Links ]
8. Sidiqi MH, Gertz MA. Daratumumab for the treatment of AL amyloidosis. Leuk Lymphoma [Internet]. 2019;60(2):295–301. Available from: https://doi.org/10.1080/10428194.2018.1485914 [ Links ]
9. Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, et al. Open Access AL Amyloidosis. 2012;1–13. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3495844/pdf/1750-1172-7-54.pdf [ Links ]
10. Vidal-Crespo A, Matas-Céspedes A, Rodriguez V, Rossi C, Valero JG, Serrat N, et al. Daratumumab displays in vitro and in vivo anti-tumor activity in models of B cell non-Hodgkin lymphoma and improves responses to standard chemo-immunotherapy regimens. Haematologica. 2019;haematol. 2018.211904. [ Links ]
11. Plesner T, Krejcik J. Daratumumab for the treatment of multiple myeloma. Front Immunol. 2018;9(JUN):1–10. [ Links ]
12. Hossein Taghizadeh MA, Thomas R, et al. Daratumumab – a safe first-line treatment of cardiac AL amyloidosis in heavily compromised patients. The XVIth International Symposium on Amyloidosis; March 26–29, 2018; Kumamoto, Japan: International Society of Amyloidosis; 2018. [ Links ]
13. Arnall JR, Usmani SZ, Adamu H, Mishkin J, Bhutani M. Daratumumab, pomalidomide, and dexamethasone as a bridging therapy to autologous stem cell transplantation in a case of systemic light-chain amyloidosis with advanced cardiac involvement. J Oncol Pharm Pract. 2019;25(4):1021–1025. [ Links ]
14. Palladini G, Kastritis E, Maurer MS, Zonder J, Minnema MC, Wechalekar AD, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136(1):71–80. [ Links ]
Clara Pardinhas, MD
Nephrology Department, Coimbra University Hospital Center
Faculty of Medicine of the University of Coimbra
Praceta, R. Prof. Mota Pinto, 3004-561 Coimbra
E-mail: clarapardinhas@gmail.com
Disclosure of potential conflicts of interest: none declared
Received for publication: Jul 26, 2020
Accepted in revised form: Sep 14, 2020