








Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.34 no.1 Lisboa Mar. 2020
https://doi.org/10.32932/pjnh.2020.04.066
CASE REPORT
A physiological approach to recurrent nephrolithiasis and its genetic determinants
Nuno Moreira Fonseca1, Marine Livrozet2,3, Rosa Varga-Poussou4, Emmanuel Letavernier2,3, Vincent Frochot2,3, Michel Daudon2,3, Jean-Philippe Haymann2,3
1 Nephrology Department, Centro Hospitalar Universitário de Lisboa Central, Hospital Curry Cabral, Lisbon, Portugal.
2 Service d'Explorations Fonctionnelles Multidisciplinaires, AP-HP, Hôpital Tenon, Paris, France
3 Sorbonne Université, UMR-S 1155, GRC n°20, Paris, France
4 Département de Génétique, Hôpital Européen Georges Pompidou, AP-HP, Paris, France
ABSTRACT
We report a case of a 63-year-old patient with recurrent nephrolithiasis for over 40 years and a significant family history of nephrolithiasis.
The patient underwent full investigation at our department. He presented hypercalcemia, hypophosphatemia and hypercalciuria, with parathyroid hormone level in the normal range. A calcium load test and a fluorocholine PET-CT excluded primary hyperparathyroidism. Abnormal secretion of parathyroid hormone-related protein and sarcoidosis were also excluded. Genetic analysis showed mutations encoding for 25(OH)-vitamin D3-24-hydroxylase (CYP24A1) and Na-dependent phosphate cotransporter 2c (SLC34A3). This case affords insights into the biological pathways that underlie the role of genetic inheritance and accrued risk of development of nephrolithiasis.
Keywords: Calcitriol, Hypercalciuria, Hyperphosphaturia, Nephrolithiasis
INTRODUCTION
The formation of kidney stones stems from a wide range of underlying disorders. It is relevant for clinicians to look for underlying causes of nephrolithiasis to direct medical management. Substantial progress has been made in identification of metabolic risk factors predisposing nephrolithiasis. The role of specific genetic and epigenetic factors has remained less clear. Although important advances have been made in understanding nephrolithiasis due to single gene defects, the understanding of polygenetic causes is still elusive.1 Until recently familial clustering of idiopathic nephrolithiasis was the main support of a suggested genetic basis. Genetic testing for the most common idiopathic forms of nephrolithiasis has yielded a number of promising candidate genes, such as SPP1 (osteopontin), CASR (calcium-sensing receptor), VDR (vitamin D receptor), CLDN14 (claudin 14).2,3 Timely diagnosis of genetic determinants of nephrolithiasis is warranted to prevent development of kidney injury; however, diagnosis is often delayed owing to unfamiliarity with these rare disorders.4 Advances in the management of nephrolithiasis depend on the combined efforts of clinicians and scientists to understand the pathophysiology.
CASE REPORT
A 63-year-old man with bilateral recurrent kidney stones was referred to our department. Nephrolithiasis onset occurred at age 13 and the patient presented more than 20 episodes over his lifetime.
He underwent left percutaneous nephrolithotomy over 20 years ago. Remaining stones were spontaneously expelled. None of his kidney stones have been analyzed. His medical history was unremarkable except for recent onset hypertension treated with irbesartan and a traumatic humerus fracture 2 years before. Of note, he had a family history of nephrolithiasis with 3 out of 5 of his siblings presenting kidney stones. He did not take any medication that interfered with calcium or phosphate homeostasis. His daily water intake was 1.5 liters and he had stopped consumption of dairy products in the last 2 years. His physical examination findings were normal. Renal ultrasound showed 2 right side kidney stones (5-6mm).
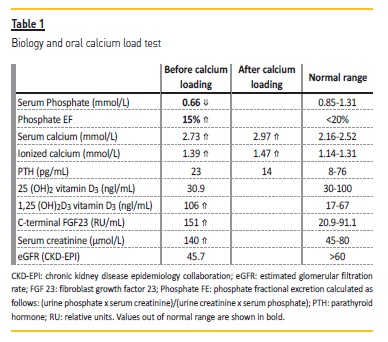
Biochemistry revealed an increased serum ionized calcium level of 1.39 mmol/L (1.14<N<1.31), low serum phosphate level at 0.7mmol/l (0.85<N<1.31), a parathyroid hormone (PTH) level in the normal range at 32 pg/mL (8<N<76), and an elevated calcitriol serum level of 106 pg/ml (17<N<67). Urine calcium excretion was increased at 10.3 mmol/24h. Fasting urine pH was 6.2. An oral calcium load test was performed to investigate hypercalciuria and results are presented in Table 1. A fluorocholine PET-CT showing normal parathyroid glands without hypermetabolic foci, ruled out primary hyperparathyroidism (PHPT) and a thoracic-abdominal-pelvic CT scan did not show any findings suggestive of granulomatous lesions. Furthermore, PTHrP and angiotensin-converting enzyme levels were normal. Testing for CYP24A1, SLC34A1, SLC34A3, SLC9A3R1 genes was positive for a homozygous CYP24A1 mutation (c.1186C>T; p.Arg396Trp) and a SLC34A3 variant (c.1357_1359del; p.DelPhe453).
DISCUSSION
As the patient presented hypercalcemia, hypercalciuria, and renal phosphate loss, PHPT could be responsible for his condition. In the presence of PHPT, a calcium load test will increase serum calcium while PTH secretion should not be adequately decreased. In our patient, calcium load test showed further increase in serum calcium as expected. However, it was unclear whether the degree of PTH level decrease was appropriate to rule out PHPT. For this reason, we decided to further evaluate the hypothesis of PHPT by performing a fluorocholine PET-CT to evaluate the parathyroid glands. Imaging ruled out PHPT. Normal serum levels of parathyroid hormone-related protein (PTHrP, < 8.5 pg/mL) ruled out a PTHrP-secreting tumor.
Our patient also presented increased levels of calcitriol. In the absence of calcitriol supplementation, elevated calcitriol levels can be due to increased production or diminished degradation. The former occurs in the setting of increased 1-alpha-hydroxylase activity, which catalyzes the conversion of 25 (OH)2 vitamin D3 into calcitriol. 1-alphahydroxylase (CYP27B1) is elevated in the setting of sarcoidosis due to increased production in macrophages. Increased calcitriol levels are also seen in the presence of reduced activity of 25(OH)-vitamin D3-24-hydroxylase (CYP24A1), the enzyme that promotes calcitriol degradation. Regarding increased activity of 1-alpha-hydroxylase, our patient did not present any signs or symptoms of granulomatous disease.
Moreover, thoracic-abdominal-pelvic CT scan and angiotensinconverting enzyme levels were normal.
Surprisingly, despite elevated levels of calcitriol, phosphatemia was decreased due to renal phosphate wasting (phosphate EF was elevated). Hence we hypothesized that the occurrence of both disturbances of renal phosphate wasting and hypercalcitriolemia could be explained by underlying genetic mutations affecting genes coding for renal phosphate handling and enzymes acting on vitamin D metabolism.
Gene sequencing showed a homozygote mutation CYP24A1 and a heterozygous variant of SLC34A3 genes, respectively encoding for 25(OH)-vitamin D3-24-hydroxylase and Na-dependent phosphate cotransporter 2c. The CYP24A1 mutation identified in our patient DNA (exon 9; homozygous variation; c.1186C>T; p.Arg396Trp; allele frequency <1/1,500; missense; considered pathogenic – American College of Medical Genetics (ACMG) 2015, class 5) has been previously described.5 Bi-allelic CYP24A1 mutations can cause idiopathic infantile hypercalcemia, characterized by failure to thrive and nephrocalcinosis, typically presenting in the first year of life, as well as a more rare late onset presentation with hypercalcemia-induced nephrolithiasis generally leading to advanced chronic kidney disease.6 Our patient's history of recurrent nephrolithiasis was reported at the age of 13 years, which is suggestive of heterogeneity of severity of modification in CYP24A1 activity and an extended phenotypic spectrum.7
There is an increased incidence of nephrolithiasis for heterozygous carriers of SLC34A3 mutations.8 Heterozygosity for the SLC34A3 variant (exon 13; heterozygous variation; c.1454G>A, p.(Arg485His); allele frequency 0.27% (GNOMAD); missense; likely pathogenic – ACMG 2015, class 4); found in our patient is predicted to be deleterious according to the pathogenicity prediction system UMD-Predictor Alamut. However, many carriers are asymptomatic. We have previously described another patient with kidney stones, hypercalcemia, hypercalciuria, renal phosphate leak, and intermediate PTH levels who presented both mutations, although in the previous case the mutation of CYP24A1 was present in heterozygous state.8 This additional report reiterates our hypothesis of synergic deleterious interaction between CYP24A1 and SLC34A3 mutations favours a pro-lithogenic environment. SLC34A3 mutations alone, induce defective function of the Na-dependent phosphate cotransporter 2c, leading to renal phosphate wasting and a subsequent moderate increase of calcitriol levels.9 Additionally, reduced degradation o calcitriol due to the CYP24A1 inactive mutation will further increase already elevated calcitriol levels. These subsequent dysregulations of vitamin D metabolism are thus responsible for kidney stones in the setting of a PTH independent hypercalcemia – hypercalciuria phenotype.9
We also noted that this patient presented high levels of FGF-23. In chronic kidney disease (CKD) FGF-23 levels rise in parallel with declining renal function. Our patient presented proportional levels of FGF-23 with his degree of renal failure (100<N<225 RU/mL in CKD stage 3).10 FGF-23 regulates vitamin D metabolism by inhibiting expression of CYP27B1 (the gene encoding 1-alpha-hydroxylase) and promoting expression of CYP24A1. Therefore increased FGF-23 results in decreased calcitriol levels.11 In this case, the increased level of FGF-23 may exert a mild counterbalance to CYP24A1 and SLC34A3 mutations effects on calcitriol synthesis but increases renal phosphate leak. This case afforded insights into biological pathways that appear to underlie how genetic inheritance can affect multiple checkpoints and contribute to accrued risk of development of nephrolithiasis.
References
1. Moe OW. Kidney stones: pathophysiology and medical management. Lancet. 2006;367(9507):333-344. [ Links ]
2, Taguchi K, Yasui T, Milliner DS, Hoppe B, Chi T. Genetic risk factors for idiopathic urolithiasis: A systematic review of the literature and causal network analysis. Eur Urol Focus. 2017; 3 (1): 72-81. [ Links ]
3. Monico CG, Milliner DS. Genetic determinants of urolithiasis. Nat Rev Nephrol. 2012;8(3):151. [ Links ]
4. Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, Palsson R. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013;28(10):1923-1942. [ Links ]
5. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, Misselwitz J, Klaus G, Kuwertz-Bröking E, Fehrenbach H, Wingen AM. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. 2011;365(5):410-421. [ Links ]
6. Cools M, Goemaere S, Baetens D, Raes A, Desloovere A, Kaufman JM, De Schepper J, Jans I, Vanderschueren D, Billen J, De Baere E. Calcium and bone homeostasis in heterozygous carriers of CYP24A1 mutations: A cross-sectional study. Bone. 2015;81:89-96. [ Links ]
7. Wolf P, Müller-Sacherer T, Baumgartner-Parzer S, Winhofer Y, Kroo J, Gessl A, Luger A, Krebs M. A case of late-onset idiopathic infantile hypercalcemia secondary to mutations in the CYP24A1 gene. Endocr Pract. 2014;20(5):e91-95. [ Links ]
8. Tabibzadeh N, Cheddani L, Daudon M, Haymann JP, Toussaint A, Silve C, Letavernier E. The Case| Epistasis and urolithiasis. Kidney Int. 2017;92(2):523-524. [ Links ]
9. Dasgupta D, Wee MJ, Reyes M, Li Y, Simm PJ, Sharma A, Schlingmann KP, Janner M, Biggin A, Lazier J, Gessner M. Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J Am Soc Nephrol. 2014;25(10):2366-2375. [ Links ]
10. Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012;82(7):737-747. [ Links ]
11. Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Rev. 2012;92(1):131-155.
Nuno Moreira Fonseca, MD, MA.
Department of Nephrology, Centro Hospitalar Universitário de Lisboa Central
Rua da Beneficência 8, 1069-166 Lisbon, Portugal.
E-mail: nuno.mf@nyu.edu
Disclosure ofpotential conflicts of interest: none declared
Received for publication: Nov 13, 2019
Accepted in revised form: Jan 9, 2020