








Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.33 no.1 Lisboa Mar. 2019
https://doi.org/10.32932/pjnh.2019.04.005
ORIGINAL ARTICLE
Nephrocalcinosis in a portuguese pediatric population
Sofia Bota1, Joana V. Andrade2, Telma Francisco1, Raquel Santos1, Gisela Neto1, Margarida Abranches1
1 Pediatric Nephrology Unit, Hospital de Dona Estefânia, Centro Hospitalar Universitário de Lisboa Central, Lisbon, Portugal
2 Centro Hospitalar Tondela-Viseu, EPE, Viseu, Portugal
ABSTRACT
Introduction and Aims: Nephrocalcinosis is characterized by the deposition of calcium in the kidney parenchyma and tubules. The renal prognosis depends on the underlying cause, emphasizing the importance of its identification. We aim to review the data of children with nephrocalcinosis concerning etiology, clinical manifestations, growth and renal function at presentation and outcomes.
Methods: Retrospective study of the records of children (<18 years) with nephrocalcinosis followed by a pediatric nephrology unit of level III hospital, between 2008‑17. Clinical features, etiology, treatment and outcomes were evaluated.
Results: We identified 35 cases: 24 isolated (69%) and 11 with nephrolithiasis (31%). The group was mostly constituted of girls (54%). Median age at presentation was 6 years (7 months – 17 years old); 40% of patients were under 2 years of age, 31% between 3 and 9 years and 29% older than 10 years. Mean follow‑up was 4 years (1–9). The most common clinical manifestation was failure to thrive in the first year of life (34%) and flank or abdominal pain (20%); in 23% it was an incidental finding. Eleven percent of patients had a systemic syndromic disease. Renal function at diagnosis was normal in all children. The most frequent causes were metabolic abnormalities (23%), hereditary tubulopathies (23%), prematurity (20%) and pharmacologic (14%). Eleven percent were considered idiopathic. In a logistic regression analysis, sex, age of presentation and familiar history of nephrocalcinosis showed no correlation with nephrocalcinosis, nephrocalcinosis and nephrolithiasis or hereditary/metabolic etiologies.
Discussion: Despite the small sample, in this study, the hereditary and/or metabolic disorders were the main cause of nephrocalcinosis. Associated symptoms and comorbidities, such as prematurity, growth retardation, intestinal malabsorption, or bone demineralization, should be evaluated for diagnostic purposes. No patient developed chronic kidney disease.
Keywords: nephrocalcinosis, nephrolithiasis, children, Portuguese
INTRODUCTION
Nephrocalcinosis (NC) is characterized by the deposition of calcium in the kidney parenchyma and tubules1. It is usually asymptomatic, especially during infancy and early childhood and diagnosed incidentally when ultrasound is performed for other reasons1. Reported clinical symptoms include renal colic in infants, gross or microscopic hematuria and/or sterile leukocyturia, mainly when associated to nephrolithiasis. Also, it is not unusual for NC to be diagnosed during systematic renal ultrasound examination of high‑risk infants or as part of the diagnostic evaluation of urinary tract infection1.
Increased urinary calcium excretion, with or without hypercalcemia, is the most common cause of NC, explaining its frequent association with nephrolithiasis. Equally so, nephrolithiasis often occurs in the absence of NC2. Recognized risk factors include genetic abnormalities in epithelial transport, metabolic disturbances, anatomical abnormalities, prematurity and urinary tract infections in the majority of pediatric patients1.
The physiological mechanisms of preventing crystal formation and adhesion can be foiled by high amounts of a solute due to hyperabsorption (e.g., in vitamin A/D excess, chronic inflammatory bowel disease, small bowel syndrome), overproduction (e.g., primary hyperoxaluria), deranged epithelium (e.g., infection, prematurity), and tubular transport defects2. Hypercalciuria induced by drugs such as loop diuretics, excessive calcium or vitamin D supplementation are also pointed out as a NC cause1.
Preterm neonates are at increased risk of NC as nephrogenesis is not completed until 34 to 36 weeks gestation. In addition, this risk increases with decreasing birth weight3. New and proliferating epithelium tubule cells are thought to be more susceptible to crystal formation and aggregation, particularly in the setting of tubular fluid supersaturation, which is promoted by hypercalciuria and hypocitraturia2,3. Hypercalciuria can be caused by hypercalcemia4 (associated with increased calcium and phosphate intake, especially in preterm infants receiving total parenteral nutrition, excessive vitamin D supplementation, subcutaneous fat necrosis, and typically reported genetic disorders like Williams‑Beuren syndrome) or due to decreased calcium renal reabsorption (loop diuretics5, aminoglycosides6, and tubular disorders like Bartter syndrome and distal renal tubular acidosis). Very low birth weight preterm infants also have decreased bicarbonate reabsorptive capacity that enhances calcium phosphate precipitation3.
Renal prognosis is determined by the underlying cause, which enhances the importance of its identification. Our study aims were to review the clinical presentation, causes, evolution and treatment of children with NC followed by a Pediatric Nephrology Unit.
SUBJECTS AND METHODS
Study population
Retrospective review of medical records from patients under 18 years old, referred to the outpatient clinic of a level III Hospital, from January 1st, 2008 to December 31st, 2017 with NC diagnosis.
Clinical features, past medical conditions, etiology, treatment and outcomes were evaluated. Renal lithiasis family history was assessed through a verbal questionnaire and, when available, renal ultrasonography and lithiasis composition were considered. Excessive dairy products, animal protein, oxalates, phytates and sodium were considered dietary errors, and also reduced water, fruit and vegetables intake. Dietary errors were not considered as a cause of NC, but a contributor factor.
NC diagnosis was always obtained by ultrasonography. NC etiologies were classified as metabolic (idiopathic hypercalciuria, idiopathic hyperoxaluria), hereditary tubulopathy, infectious, prematurity, pharmacologic, structural (stasis), systemic syndromic disease and idiopathic.
Workup
Blood serum with creatinine, urea nitrogen, uric acid, acid‑base status, sodium, potassium, chlorine, magnesium, phosphorus and urine analysis were performed at the first medical appointment. An evaluation of the urinary excretion was performed on 24‑hour urine samples or by random urine measurement. We considered: hypercalciuria >4 mg/kg/day, hyperoxaluria >0.5 mmol (45 mg)/1.73m2/day, hyperuricosuria >815mg/1.73m2/day, cystinuria >75mg/1.73m2/day, hypocitraturia <320 mg/1.73m2/day, hypomagnesuria <2 mmol/day. Lithogenic risk was defined as urinary calcium/citrate ratio >0.33 8‑11. Abnormal urinary excretion results were always confirmed in at least two samples.
Statistical analysis
The collected data was analyzed by SPSS, 20th version software (SPSS, Chicago, IL) for Mac using Students T test, the Mann‑Whitney U test, chi‑square test, Fishers exact test and logistic regression analysis, with P values of <0.05 considered to be significant.
Ethics
The procedures were in accordance with the ethical standards of the committee in charge of human experimentation (institutional and national) and with the Helsinki Declaration of 1975 (revised in 2015).
RESULTS
We identified 35 cases with NC: 24 isolated (69%) and 11 with nephrolithiasis (31%).
Demographic characteristics
Median age at presentation was 6 years (7 months‑old–17 years‑old) with a (2–9) interquartile range 40% under 2 years old (n=14), 31% between 2–9 years (n=11) and 29% of 10 or more years old (n=10). The proportion of female subjects was 54% (n=19). The mean follow‑up was 4 years (1–9).
Presenting symptoms at diagnosis
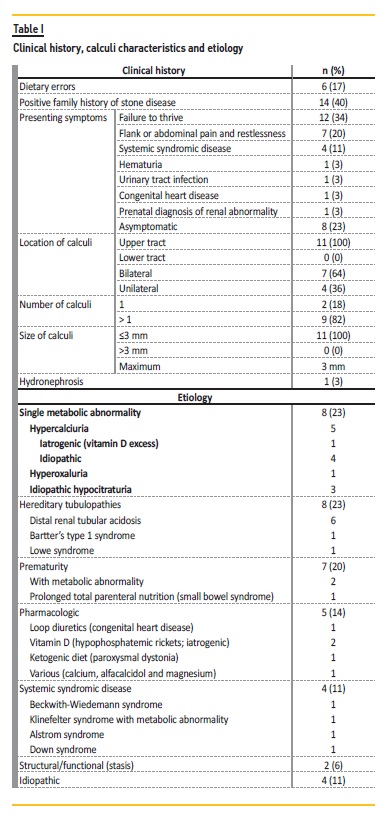
The most common finding was failure to thrive in the first year of life in 12 patients (34%): five patients were preterm neonates, five had a distal renal tubular acidosis, one had Alstrom syndrome, and one patient had a familial hypercalciuria history (waiting for genetic study result). Flank or abdominal pain was the second most reported symptom in seven cases (20%) and four of these had nephrolithiasis observed by ultrasonography. Eight patients (23%) were asymptomatic. Renal function at diagnosis was normal in all children. No patient presented hypertension and three patients had polyuria. When associated with nephrolithiasis (n=11), the upper urinary tract was the only affected site (100%, n=11) (Table I).
Etiology
The most frequent causes of NC were metabolic abnormalities (23%, n=8; Table I), hereditary tubulopathies (23%, n=8; Table I), prematurity and diuretic or parenteral feeding use (20%, n=7). In five cases (14%, n=5), treatment led to NC (iatrogenic). In two preterm infants, there was a metabolic abnormality association. In four cases (11%) there was no identifiable cause (Table I) and none had an infectious cause.
Fourteen patients (40%) had a positive nephrolithiasis family history and two (6%) had first‑degree relatives with NC. Next‑generation sequencing was performed in three siblings that are being followed for hypercalciuria, but is still pending. In a logistic regression analysis, sex, age of presentation and familiar history of NC showed no correlation with NC alone, NC associated with nephrolithiasis or hereditary/metabolic etiology.
Twenty‑four‑hour urine collection was performed in 63% (n=22). In 23% (n=8), urine evaluation revealed a metabolic disturbance (Table II) and all had a single disturbance. Globally, the most common metabolic abnormality was hypercalciuria. Lithogenic risk was present in four cases.
Within the group with associated urolithiasis (n=11), the etiology was metabolic abnormality (n=3), prematurity (n=3), hereditary tubulopathy (n=2), structural renal stasis (n=1), vitamin D supplementation (n=1) and no identifiable cause (n=1).
Dietary errors and urinary output
Dietary errors were identified in 17% (n=6): reduced water intake (n=4), excessive dairy products (n=2), reduced fruit and vegetables (n=1) and excessive salt (n=1). Of the 19 patients evaluated for urinary output, we found that 23% (n=8) had low urine output (<1 ml/kg/h).
Treatment and outcome
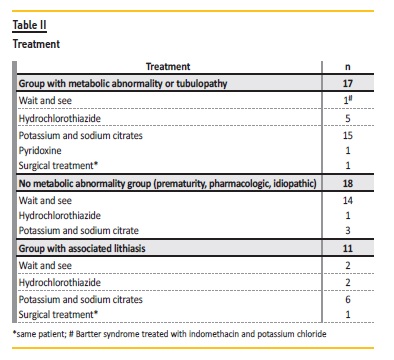
The majority was conservatively treated (97%, n=34) (Table II). Surgical treatment with ureteral stent placement was performed in one distal renal tubular acidosis case that had unilateral urolithiasis with hydronephrosis. In this case, an autosomal dominant form is more likely (first degree relative with NC and DRTA) but genetic testing is still pending.
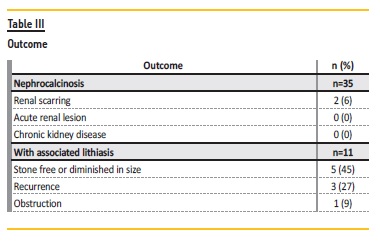
During follow‑up, complications were observed: renal scarring in 6% (n=2) and obstruction in 3% (n=1). In the group associated with nephrolithiasis, 45% (n=5) became stone free or diminished in size and 27% (n=3) had recurrence (Table III). No patient developed chronic kidney disease.
DISCUSSION
Epidemiology, sex and age
NC is an uncommon finding and its prevalence is not known. Moreover, there is a paucity of studies regarding its presentation, etiology and evolution beyond the neonatal age. NC can affect all ages, although it primarily appears to onset in the first years of life, which was also observed in our cohort. Also, we highlight that one fifth were preterm neonates, a special risk group seen in several studies12,13.
Clinical presentation
NC is classically seen as an unexpected imaging diagnosis in asymptomatic patients and usually symptomatic when associated to nephrolithiasis1.
In our study, a high proportion of patients (34%, n=12) presented failure to thrive in the first year of life, considered a clinical sign that should be investigated, as it can encompass a wide spectrum of causes. In other studies14,15, 55% to 68% of patients also presented with failure to thrive in the first year. We believe that our finding may be in relation to the young age of our cohort (40% under two years old) and to prematurity (five of 12 patients). Nevertheless, it is noteworthy that in five cases, it was this sign that prompted the initial investigation, which included a renal ultrasound that led to the renal investigation and subsequent tubulopathy diagnosis.
Etiology
NC is a non‑specific ultrasound diagnosis that always requires investigation, although there is a wide range of causes, including idiopathic cases16. Our cohort illustrates the different NC etiologies but, because of our low number of patients, we found no correlation between NC and hereditary or metabolic etiologies. Compared to Ammenti et al.14 similar sample size, only 23% of our patients had a hereditary tubulopathy and the most frequent metabolic abnormality was hypercalciuria.
Our results show an overall rate of syndromic cases (11%) comparable to the cited author14. Renal abnormalities involving the medulla and the collecting system occur in 15% to 25% of patients with Beckwith‑Wiedemmann syndrome17,18. Moreover, hypercalciuria is present in 10 to 22% of series19,20 and it can predispose to NC, also described in these patients. NC has also been reported in Klinefelter21, Alstrom syndromes22, and anecdotal cases of Down syndrome with prolonged excessive calcium intake23,24. However, in our patient no metabolic abnormality, medication or supplementation was identified.
Treatment and outcome
Therapy is directed at the underlying cause. For idiopathic cases, measures may be undertaken to reduce the urinary concentration and increase the solubility of the substances (calcium, phosphate or oxalate). Data that support such interventions are extrapolated from studies of patients with nephrolithiasis; no studies have demonstrated a beneficial effect among patients with established NC.
Unlike other authors subjects25, all our patients had a normal renal function at presentation. Furthermore, it remained stable, as was the case in the Ammenti et al. study14, that had a similar mean follow‑up time.
The renal prognosis of NC is determined by the underlying cause and the majority do not progress to end‑stage renal disease. Patients with medullary sponge kidney or distal renal tubular acidosis rarely develop chronic kidney disease, unlike underlying causes, when not effectively treated, such as primary hyperoxaluria, hypomagnesemic hypercalciuric NC and Dents disease, not identified in our group.
Regarding ultrasound findings, we observed no NC worsening even in the tubulopathies group. Moreover, we observed a nephrolithiasis regression in the majority of this subgroup of patients, which was expectable under directed therapy.
Limitations and conclusion
The major limitations of this study are the small sample size and its retrospective nature that implicates different follow‑up timings. Also, dietary intakes were estimated and not evaluated through a validated questionnaire. Still, to the best of our knowledge it is the first Portuguese study to review NC cases. Despite the small sample, we verified that hereditary and/or metabolic disorders were the main cause of NC. Associated symptoms and comorbidities, such as prematurity, growth retardation, intestinal malabsorption, or bone demineralization should be evaluated for diagnostic purposes.
Since these thresholds and limits are shared by most of the clinical settings to which patients are referred, the data presented in this study may set the stage for a prospective study and for the creation of a national database that would be fundamental for a better clinical and epidemiological understanding of these uncommon cases, particularly when idiopathic. In addition, it would be interesting to investigate possible genetic causes, in light of the recent reports of monogenic disorders associated with hypercalciuric nephrolithiasis with NC26.
References
1. Alon US. Nephrocalcinosis. Curr Opin Pediatr 1997;9(2):160–165. [ Links ]
2. Sayer JA, Carr G, Simmons NL. Nephrocalcinosis: molecular insights into calcium precipitation within the kidney. Clin Sci 2004;106(6):549. [ Links ]
3. Lee HS, Sung IK, Kim SJ, Youn YA, Lee JY, Lim GY, et al. Risk Factors Associated with Nephrocalcinosis in Preterm Infants. Am J Perinatol 2014;31:279–286. [ Links ]
4. Schell‑Feith EA, Kist‑van Holthe JE, Conneman N, van Zwieten PH, Holscher HC, Zonderland HM, et al. Etiology of nephrocalcinosis in preterm neonates: association of nutritional intake and urinary parameters. Kidney Int 2000;58(5):2102–2110. [ Links ]
5. Gimpel C, Krause A, Franck P, Krueger M, von Schnakenburg C. Exposure to furosemide as the strongest risk factor for nephrocalcinosis in preterm infants. Pediatr Int 2010;52:51–56. [ Links ]
6. Narendra A, White MP, Rolton HA, Alloub ZI, Wilkinson G, McColl JH, et al. Nephrocalcinosis in preterm babies. Arch Dis Child Fetal Neonatal Ed 2001;85:207–213. [ Links ]
7. López M, Hoppe B. History, epidemiology and regional diversities of urolithiasis. Pediatr Nephrol 2008; 25: 49–59. [ Links ]
8. Cameron MA, Sakhaee K, Moe OW. Nephrolithiasis in children. Pediatr Nephrol 2005;20:1587–1592. [ Links ]
9. Saez‑Torres C, Grases F, Rodrigo D, Garcia‑Raja AM et al. Risk factors for urinary stones in healthy schoolchildren with and without a family history of nephrolithiasis. Pediatr Nephrol 2013;28:639–645. [ Links ]
10. Gürgöze MK, Sarı MY. Results of medical treatment and metabolic risk factors in children with urolithiasis. Pediatr Nephrol 2011;26:933–937. [ Links ]
11. Oguz U, Resorlu B, Unsal A. Metabolic evaluation of patients with urinary system stone disease: a research of pediatric and adult patients. Int Urol Nephrol 2014;46:329–334. [ Links ]
12. Narendra A, White MP, Rolton HA, Alloub ZI, Wilkinson G, McColl JH, Beattie J. Nephrocalcinosis in preterm babies. Arch Dis Child Fetal Neonatal Ed 2001;85:F207–F213. [ Links ]
13. Schell‑Feith EA, Kist‑van Holthe JE, van der Heijden AJ. Nephrocalcinosis in preterm neonates. Pediatr Nephrol 2010;25(2):221–230. [ Links ]
14. Ammenti A, Pelizzoni A, Cecconi M. Nephrocalcinosis in children: a retrospective multi‑centre study. ActaPaediatr 2009;98(10):1628–1631. [ Links ]
15. Al‑Bderat JT, Mardinie RI, Salaita GM, Al‑Bderat AT, Farrah MK. Nephrocalcinosis among children at king hussein medical center: causes and outcome. Saudi J Kidney Dis Transpl 2017;28(5):1064–1068. [ Links ]
16. Arthurs O, Easty M, Riccabona MM. Grainger & Allisons diagnostic radiology, 6th edition. Imaging of the kidneys, urinary tract and pelvis in children. Elsevier Limited. 2015;78:1848–1890.e11.
17. Choyke PL, Sigel MJ, Oz O,Sotelo‑Avila C, De Baun MR. Non malignant renal disease in pediatric patients with Beckwith‑Weidemann syndrome. Am J Radiol 1998;171:733–737. [ Links ]
18. Goldman M, Smith A, Shuman C, Caluseriu O, Wei C, Steele L, et al. Renal abnormalities in Beckwith‑Wiedemann syndrome are associated with 11p15.5 uniparental disomy. J Am Soc Nephrol 2002;13:2077–2084. [ Links ]
19. Mussa A, Peruzzi L, Chiesa N, De Crescenzo A, Russo S, Melis D, et al. Nephrological findings and genotype‑phenotype correlation in Beckwith‑Wiedemann syndrome. Pediatr Nephrol 2012;27(3):397–406. [ Links ]
20. Goldman M, Shuman C, Weksberg R, Rosenblum ND. Hypercalciuria in Beckwith‑Wiedemann Syndrome. J Pediatr 2003;142(2):206–208. [ Links ]
21. Nahata L, Yu RN, Paltiel HJ, Chow JS, Logvinenko T, Rosoklija I, et al. Sperm Retrieval in Adolescents and Young Adults with Klinefelter Syndrome: A Prospective, Pilot Study. J Pediatr. 2016 Mar;170:260–5.e1‑2 [ Links ]
22. Baig S, Paisey R, Dawson C, Barrett T, Maffei P, Hodson J, et al. Defining renal phenotype in Alström syndrome. Nephrol Dial Transplant 2018; doi: 10.1093/ndt/gfy293 [ Links ]
23. Proesmans W, De Cock P, Eyskens B. A toddler with Down syndrome, hypercalcaemia, hypercalciuria, medullary nephrocalcinosis and renal failure. Pediatr Nephrol 1995;9:112–114. [ Links ]
24. Ramage IJ, Durkan A, Walker K, Beattie TJ. Hypercalcaemia in association with trisomy 21 (Downs syndrome). J Clin Pathol 2002;55(7):543–544. [ Links ]
25. Rönnefarth G, Misselwitz J. Nephrocalcinosis in children: a retrospective survey. Members of the Arbeitsgemeinschaft für pädiatrische Nephrologie. Pediatr Nephrol. 2000;14(10‑11): 1016–1021. [ Links ]
26. Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol 2015;26(3):543–551. [ Links ]
Sofia Bota, MD
Rua Jacinta Marto, 1169‑045, Lisboa
Disclosure of potential conflicts of interest: none declared.
Received for publication: Mar 1, 2018
Accepted in revised form: Mar 20, 2019