








Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.4 Lisboa Dec. 2018
REVIEW ARTICLE
Heart failure in chronic kidney disease patients: a summary of non-traditional risk factors
Luísa Viveiros1, Manuel Pestana2
1 Faculty of Medicine of Porto University, Porto, Portugal
2 Division of Nephrology, São João Hospital Centre and Department of Medicine, Faculty of Medicine of Porto University, Porto, Portugal
ABSTRACT
Introduction: Patients with chronic kidney disease are at higher risk of heart failure than the general population. Such increased risk is multifactorial, being partly due to a higher prevalence of traditional cardiovascular risk factors in this population and to the existence of non-traditional/chronic kidney disease-specific risk factors. Non-traditional risk factors should prompt special considerations in diagnosis and management of heart failure in these patients.
Material and methods: We searched PubMed and Medline during 2017 and 2018, for the terms Chronic kidney disease and Heart failure. Other search terms were included according to the content found. We mainly focused on publications concerning chronic kidney disease-specific risk factors for heart failure and its pathophysiology. Further publications regarding management of heart failure in chronic kidney disease patients were sought.
Results and discussion: Heart failure is an important cardiovascular complication in chronic kidney disease. Specific risk factors for heart failure, which are not often found in the general population, include, among others, mineral bone disease, uremic toxins, neuro-hormonal activation, inflammation, oxidative stress, anemia, malnutrition, albuminuria and hyperhomocysteinemia. Management of heart failure in this group of patients is challenging and patients are often undertreated.
Conclusion: Research into non-traditional risk factors for heart failure in chronic kidney disease patients is lacking and its pathophysiology is not properly understood. Such understanding is vital to provide proper care, as in the general population. We also question whether some of these specific risk factors could also play a role in heart failure among the general population.
Keywords: Cardiovascular risk factors; chronic kidney disease; heart failure.
INTRODUCTION
Chronic kidney disease (CKD) is a major public health problem, affecting more than 10% of the general population in many countries worldwide.1
The first report of an association between CKD and cardiovascular abnormalities goes back to 1836.2 Subsequently, several studies in this area have been performed and the relationship between CKD and increased cardiovascular risk is well established.
Cardiovascular disease and kidney disease are closely interrelated and disease of one organ causes dysfunction of the other, ultimately leading to the failure of both organs. This is often referred as cardiorenal syndrome (Table I).3

In CKD patients, heart failure (HF) is an important cardiovascular complication4 and an important cause of morbidity and mortality.5 Chronic kidney diseasespecific complications contribute to vasculopathy and cardiomyopathy in these patients, ultimately leading to HF through unique pathophysiologic mechanisms, distinct from the ones contributing to HF in patients without CKD. Therefore, HF in CKD patients requires special considerations regarding diagnosis and management, in comparison to HF in patients without CKD.
Despite the important impact of HF in CKD patients prognosis, the predictors of HF as a specific end point have not been satisfactorily characterized in subjects with CKD.5
In this review, we address the unique non-traditional/ CKD-specific risk factors for HF in CKD patients and their implications in diagnosis and therapy, which should be taken in account when approaching HF in CKD patients.
For this purpose, we searched PubMed and Medline during 2017 and 2018, for the terms Chronic kidney disease and Heart failure. Other search terms were included according to the content found. We mainly focused on publications concerning CKD-specific risk factors for HF and its pathophysiology. Further publications regarding management of HF in CKD patients were sought.
CHRONIC KIDNEY DISEASE
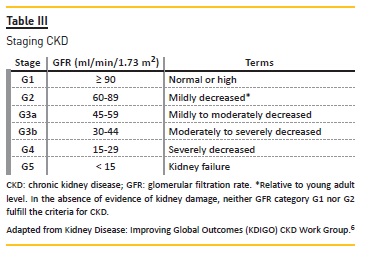
Chronic kidney disease is defined as abnormalities of kidney structure or function, present for > 3 months, with implications for health.6 Criteria and staging for CKD are presented in Tables II and III, respectively.

Chronic kidney disease is frequently associated with a progressive decrease in GFR, frequently leading to endstage renal disease (ESRD).7
Prevalence of CKD worldwide is estimated to be 8-16%. Contrary to clinically apparent advanced-stage CKD, precise calculation of the burden of less symptomatic or asymptomatic early-stages CKD is difficult. These account for 80-90% of all cases.8
In 1990, CKD was ranked as the 27th cause of total number of global deaths (age-standardized annual death rate of 15.7 per 100 000). In 2010, it rose to 18th (annual death rate of 16.3 per 100 000).9 These rates are likely underrated. Reported mortality from diabetes-related renal disease is estimated to be four to nine times less than the actual rate.10
Diabetes and hypertension are the leading causes of CKD in all developed and many developing countries. In countries of Asia and sub-Saharan Africa, glomerulonephritis and unknown causes are more common.
Genetic factors, such as variations in MYH9 and APOL1, are associated with non-diabetic CKD in Africans.8
As a result of the loss of endocrine and exocrine functions of the kidney, people with CKD are prone to developing various complications, namely vitamin D deficiency, hyperphosphatemia, hyperparathyroidism, acidosis, anemia, hypoalbuminemia and hypertension, among others.6 These lead to other complications, including bone disorders and fractures, cognitive decline, and an increase in all-cause and cardiovascular mortality.8 Patients with CKD should be considered at increased risk for cardiovascular disease.6 As mentioned, HF is an important cardiovascular complication.4
HEART FAILURE
Heart failure is defined as a clinical syndrome characterized by typical symptoms (breathlessness, ankle swelling and fatigue) that may be accompanied by signs (elevated jugular venous pressure, pulmonary crackles and peripheral edema), caused by structural and/or functional cardiac abnormalities, resulting in reduced cardiac output and/or elevated intracardiac pressures at rest or during stress.11
In developed countries, HF has an estimate prevalence of 1-2% among adults, rising to ≥ 10% in individuals > 70 years of age.11 This condition shows substantial morbidity and mortality. Indeed, it is the most frequent cause of hospital admission for people over the age of 60 years in the United States.12 Twelve-month all-cause mortality rates for hospitalized and stable/ambulatory HF patients are 17% and 7%, respectively. Twelve-month hospitalization rates are 44% and 32%, respectively.13
Heart failure is caused by a loss of a significant amount of functional myocardial cells after injury to the heart.14
The etiology is wide-ranging worldwide and there is no consensual classification system, due to common overlap between categories. Patients often present with different pathologies (cardiovascular and non-cardiovascular) underlying HF.11 The most common etiologies are ischemic heart disease, hypertension and diabetes, whereas less common causes include cardiomyopathies, infections (viral myocarditis, Chagas disease), toxins (alcohol, cytotoxic drugs), valvular disease and prolonged arrhythmias.14 The proposed classification system is divided into diseased myocardium, arrhythmias and abnormal loading conditions. Among the latter, a mechanism that can contribute to HF is CKD.11
Heart failure is described according to the measurement of left ventricular ejection fraction (LVEF). Patients with LVEF ≥ 50% present HF with preserved ejection fraction (HFpEF). Patients with LVEF < 40% present HF with reduced ejection fraction (HFrEF). Patients with LVEF between 40-49% are now defined as presenting HF with mid-range ejection fraction (HFmrEF).11 In the general population, HFpEF currently accounts for > 50% of all HF cases and its prevalence relative to HFrEF continues to rise at an alarming rate of 1% per year.15
CHRONIC KIDNEY DISEASE AND HEART FAILURE: AN INDEPENDENT ASSOCIATION
Heart failure risk is increased among CKD patients. Several studies have shown an independent association between lower glomerular filtration rate (GFR) and HF16, suggesting the existence of important non-traditional/CKD-specific risk factors for HF in CKD patients.
Beck et al observed a strong association between lower GFR and HF, which remained significant for HF after multivariable adjustment.1 In the United States, in 2016, the overall prevalence of HF in individuals aged over 66 was 28.8% in CKD patients, as opposed to 6.4% in patients without CKD.17 In the Atherosclerosis Risk in Communities study, the incidence of HF was 3-fold higher for individuals with GFR <60 mL/min/1.73m2, versus the reference group (GFR ≥ 90 mL/min/1.73m2).
The overall adjusted relative hazard of developing HF was 1.94 (1.49 to 2.53) for individuals with GFR <60 mL/min/1.73m2, versus the reference group and was significantly increased for individuals with and without prevalent coronary heart disease at baseline.18
Löfman et al found a higher prevalence of CKD in HFpEF than in HFmrEF and HFrEF (56% vs 48% and 45%, respectively, p <0.001). Hypertension, atrial fibrillation and valvular heart disease were more common in HFpEF whereas ischemic heart disease and revascularization were more common in HFmrEF and HFrEF. All these variables increased with worse kidney function. Patients with worse kidney function more often had HF with longer duration and were in a more severe class, regardless of LVEF. In addition, CKD was strongly associated with mortality in all LVEF groups, in keeping with earlier studies.19
NON-TRADITIONAL RISK FACTORS FOR HEART FAILURE IN CHRONIC
KIDNEY DISEASE PATIENTS
Heart failure and CKD frequently coexist and share common risk factors (diabetes, hypertension, hyperlipidemia) that interact to worsen prognosis.11
Multiple mechanisms contribute to the pathophysiology of HF in patients with kidney disease. Traditional HF risk factors such as age, male sex, hypertension, Diabetes mellitus, smoking, obesity, coronary artery disease, left ventricular hypertrophy (LVH) are more prevalent among CKD patients than in the general population.
These patients also show unique non-traditional/kidney-specific risk factors for HF, namely malnutrition, uremic toxins, mineral bone disturbances, anemia and sympathetic hyperactivity, among others20, that will be reviewed in this article.
CKD-MINERAL BONE DISEASE
Mineral and bone homeostasis, maintained through interactions between calcium, phosphorus, parathyroid hormone (PTH), vitamin D and fibroblast growth factor (FGF-23) is commonly disrupted in CKD patients (especially when GFR <45 ml/min/1.73 m2).21
Mineral abnormalities
CKD patients are not able to form 1,25(OH)2 vitamin D nor maintain its normal serum levels. Secondary hyperparathyroidism occurs, as a result of decreased serum calcium, increased serum phosphorus, and decreased production of calcitriol.22 Hypocalcemia and hyperphosphatemia can precipitate arrhythmias and depress cardiac contractility.23
FGF-23 is a bone-derived hormone that regulates vitamin D synthesis in renal proximal tubules and renal phosphate reabsorption.3 FGF-23 increases renal phosphate excretion, by reducing the expression of Na/PiIIa cotransporter, and decreases circulating calcitriol [1,25(OH)2D3] levels. FGF-23 levels gradually increase with declining renal function, allowing the body to maintain normal phosphate levels (2.5-4.5 mg/dl) until very advanced CKD stages.7 Increased FGF-23 levels are associated with an increased risk of all-cause mortality, cardiovascular events and progression of CKD.24 This has been suggested to be the missing link between CKD and cardiovascular disease.3
An independent association between FGF-23 and LVH has been shown. Moreover, in vitro and animal model studies demonstrated that FGF-23 directly causes pathological cardiac hypertrophy.25 FGF receptordependent activation of the calcineurin-NFAT-signaling pathway independent of klotho (FGF-23 co-receptor; only expressed in kidney and parathyroid glands) may be the basis of this association.7 Scialla et al also reported an independent association between higher FGF-23 levels and greater risk of cardiovascular events, particularly HF in CKD stages 2-4 patients.26 Similarly, in the Cardiovascular Health Study, the risk of HF had a 94% increase among CKD patients with the highest quartile of FGF-23.27 Scialla et al showed that FGF-23 was not independently associated with coronary artery calcification in patients with CKD stages 2-4.28 These results suggest that direct effects of FGF-23 on cardiac remodeling, instead of arterial vasculature, may underlie its association with mortality.7
Vascular calcification
Vascular calcification can occur, through two mechanisms. The first involves focal calcification in the intima of vessels, associated with lipid-laden foam cells found in atherosclerotic plaques. The second involves diffuse calcification in the media of vessels – Mönckebergs sclerosis – which increases the blood vessel stiffness and decreases the vessel compliance, leading to cardiovascular disease.29
Carotid-femoral pulse wave velocity (CF-PWV) is the gold-standard for assessment of large artery stiffness.5
As GFR declines, CF-PWV increases. Each 10 ml/ min/1.73 m2 decline in GFR is independently associated with a 0.23 m/s increase in CF-PVW.30 Chirinos et al demonstrated that CF-PWV is associated with a marked increase in the risk of incident HF in a CKD population.
This association persisted after adjustment for blood pressure and other confounders.5
Large artery stiffness occurs secondary to arteriosclerosis (degeneration, fibrosis and calcification) of the medial layer of the aorta.5 Consequently to increased arterial stiffness, a greater reflection of the pulse wave back to the central aorta occurs, which adds extra work to left ventricular (LV) contraction.30 Thus, large artery stiffness has an important impact on LV pulsatile afterload, affecting early aortic systolic pressure rise, the total compliance of the arterial system and the velocity at which pulse waves travel forward in the arteries and reflected waves travel backwards towards the heart.5 Furthermore, large artery stiffening is associated with lower diastolic pressure, contributing to a decreased coronary perfusion pressure.5 Such increase in the pulsatile afterload is likely the mechanism responsible for the relation between aorta stiffness and risk of incident HF, leading to increased systolic wall stress, diastolic and systolic dysfunction and LVH.5
UREMIC TOXINS
As CKD develops, parenchymal and glomerular sclerosis evolves, eventually leading to uremia.23 In CKD, uremia itself is associated with the development of LVH, due to accumulation of hypertrophic substances and insulin resistance.31
Hypertrophic substances associated with uremia include endothelin 1, PTH, tumor necrosis factor-alpha (TNF-α), leptin, interleukin (IL)-1, IL-6 and cardiotonic steroids. Ouabain and marinobufagenin, two endogenous cardiotonic steroids, interact with the α-subunit of the Na+/K+-ATPase transmembrane protein on the surface of cardiomyocytes. They are found in high concentrations in CKD patients, but their role in uremic cardiomyopathy pathogenesis is not well understood.31
Uremia is associated with underlying insulin resistance, which plays a role in uremic cardiomyopathy through the Akt pathway. Insulin displays pleiotropic and metabolic actions through intracellular signaling pathways, namely the Akt pathway. Insulin resistance in these patients leads to disruption of this pathway, which is believed to contribute to the development of LVH.31
N EURO-HORMONAL ACTIVATION: SYMPATHETIC NERVOUS SYSTEM
(SNS) AND RENIN-ANGIOTENSINALDOSTERONE SYSTEM (RAAS)
The development of CKD is associated with increased activation of the SNS, which has been suggested as an important mechanism for cardiovascular events in this population.32 Plasma catecholamine concentrations are approximately doubled in CKD patients.33,34
The kidneys have both afferent and efferent nerve fibers, which are located in the adventia around the renal artery.35 The signal that commands the brain to increase sympathetic outflow is generated in the diseased kidneys.36 Graded stimulation of efferent activity induces renin release and subsequent RAAS activation, followed by an increase in sodium reabsorption and a decrease in renal blood flow at higher levels of stimulation.35
In addition to being associated with elevated blood pressure and progression of kidney damage independently of blood pressure in CKD patients, sympathetic hyperactivity is also related to LVH and HF.36 Excessive SNS activation can cause decreased myocardial betaadrenergic receptor density (particularly β1), uncoupling of the receptor from intracellular signaling mechanisms and cardiomyocyte dysfunction and fibrosis, leading to cardiac remodeling.23 Siddiqui et al showed that left ventricular mass (LVM) in CKD patients is greater than in healthy controls, even though arterial blood pressure in the patients was reasonably well controlled for a long period of time. There was a positive relationship between LVM measurements (LVM index and LV mean wall thickness) and sympathetic activity, independent of blood pressure.37 Indexes of LVM correlated with plasma norepinephrine levels.36
M ALNUTRITION-INFLAMMATIONATHEROSCLEROSIS (MIA) SYNDROME
This CKD-related syndrome features closely related malnutrition and chronic inflammation, leading to atherosclerotic damage and increased cardiovascular mortality.23,38 Pathophysiology comprises chronic inflammation leading to increased proinflammatory cytokine synthesis, activation of protein catabolism, inhibition of albumin production in the liver and decreased muscle mass. Proinflammatory cytokines also play a role by suppressing appetite. Ultimately, these substances cause cardiovascular – and cerebrovascular – complications, by damaging the vascular endothelium.38
Inflammation
Persistent, low-grade inflammation is frequently present in CKD, and it is linked to cardiovascular disease, protein-energy wasting (PEW), and cardiovascular and all-cause mortality. An inverse correlation between GFR and inflammation has been reported.39 Patients with CKD produce multiple inflammatory cytokines, which regulate cell survival and death, such as TNF-α, transforming growth factor-beta (TGF-β), IL-1 and IL-6.23
Multiple factors may cause immune dysregulation and inflammatory activation in CKD.39
Chronic kidney disease patients present decreased clearance of pro-inflammatory cytokines. Uremic milieu produces both oxidative and carbonyl stress, which are highly pro-inflammatory. Uremic toxins may lead to intestinal dysbiosis and an increased translocation of gut bacteria and bacterial components into the circulation, contributing to systemic inflammation.39 CKD patients also display raised levels of endotoxin, possibly due to dysbiosis and fluid overload that may occur in these patients. The latter favors mesenteric venous congestion and bowel wall edema, with translocation of gram-negative bacteria trough the endothelium of the intestinal villi.40 Dialysis patients are prone to inflammatory stimulation caused by catheter-related bloodstream infections, access site infections, thrombosed arteriovenous fistulas and grafts (hemodialysis patients) and episodes of peritonitis (peritoneal dialysis patients).41 Increased neuro-hormonal activity in CKD patients may also contribute to inflammatory activity.
In animal models, angiotensin II upsurges TNF-α and IL-6 expression in cardiomyocytes, possibly through increased activation of nuclear factor kappa-light-chainenhancer of activated B cells (NF-Κβ) and activator protein 1 (AP-1).42 Also in animal models, chronic stimulation with the beta-adrenergic agonist isoproterenol led to increased mRNA synthesis of pro-inflammatory cytokines TNF-α, IL-6 and IL-1β in cardiomyocytes and cardiac blood vessels.43
In animal models, TNF-α mediates progressive LV dilation and dysfunction, as well as increased cardiac mortality.40 Both TNF-α and IL-1β reduce contraction amplitude, sarcoplasmic reticulum calcium concentration and calcium transient amplitude in rat ventricular myocytes.44 TNF-related weak inducer of apoptosis (TWEAK) has also been shown to increase cardiac apoptosis and extracellular matrix degradation, thus contributing to adverse ventricular remodeling in HF.40
Malnutrition
CKD patients frequently present malnutrition, particularly in the form of PEW. PEW occurs in about 30% of patients undergoing dialysis. In this state, there is a multifactorial decrease in body protein mass and body fat.38
Suboptimal nutritional intake is a contributive factor for malnutrition in CKD. It is frequent among CKD and ESRD patients, a direct risk for protein malnutrition.45
There is a need for careful control of dietary protein intake in CKD patients, as excess dietary protein leads to the accumulation of uremic toxins.6 However, insufficient protein intake may lead to loss of lean body mass and malnutrition, which is more common in the elderly.6 Other factors leading to malnutrition in CKD include the accumulation of uremic toxins, inflammation, oxidative stress, and insulin resistance.38
Despite being acknowledged as CKD-specific risk factor for HF, further investigation into the role of malnutrition in promoting cardiac disease is lacking.20
OXIDATIVE STRESS
Oxidative stress occurs among CKD patients as a consequence of an increase in reactive oxygen species (ROS) production and decrease in antioxidant defenses.46 Increased ROS generation in CKD patients may be prompted by inappropriate activation of the RAAS and, later on, diminished nitric oxide (NO) availability (as inactivation of NO by ROS takes place)23, uremia and impaired mitochondrial respiratory system.46
Excessive ROS lead to cellular dysfunction, protein and lipid peroxidation, DNA damage and eventually cell damage and death. Besides having direct cytotoxic effects, ROS, in particular, superoxide (O2.-), can spontaneously combine with NO to form peroxynitrite (ONOO-). This reaction culminates both with the formation of a highly reactive antioxidant and the consumption of cytoprotective NO. NO has an important role in normal heart function, as it participates in coronary vasodilatation, inhibition of platelet and neutrophil adhesion and activation and inhibition of oxidative stress.47
Oxidative stress has been growing as a pathophysiologic mechanism of cardiac remodeling, contributing to HF development and progression. ROS activate a wide range of hypertrophy signaling pathways, apoptosis inducers, inflammatory mediators and proteolytic enzymes, ultimately contributing to LVH.47
ANEMIA
Anemia is common among CKD patients.23 There is a relationship between decreased GFR and anemia. As kidney function declines and in more advanced CKD stages patients, the incidence and prevalence of anemia increase.48
In CKD patients, anemia is primarily due to insufficient erythropoietin (EPO) production.23 Erythropoietin is synthetized in the cortex and outer layer of the renal medulla, by specialized type I interstitial fibroblasts. In healthy individuals, the main stimulus for EPO synthesis is tissue hypoxia. However, pathological conditions affecting the kidney in CKD may disrupt this feedback, leading to developing anemia which is not satisfactorily compensated by an adequate increase in EPO synthesis. Another important mechanism underlying renal anemia is iron deficiency. It may occur because of stimulation of hepcidin production by proinflammatory cytokines, particularly IL-6.48 Chronic kidney disease is associated with chronic inflammation, which can induce hepcidin increase (up to 100-fold).49
Increased hepcidin levels block iron absorption from food in intestines and its release from iron stores.48 Furthermore, some degree of dilution anemia due to hypervolemia may also take place in CKD.50
Due to anemia-induced tissue ischemia, peripheral vasodilatation and lowering of the blood pressure occur. This leads to SNS activation, causing tachycardia, increased stroke volume and renal vasoconstriction, which can lead to water and salt retention. Reduced renal flow activates the RAAS and antidiuretic hormone, triggering additional renal vasoconstriction and water retention. Increased plasma volume causes ventricular dilation, which increases strain on the previously stressed heart. Ultimately, LVH develops, which can lead to apoptosis and necrosis of cardiomyocytes.51
Additionally, anemia can contribute to an anomalous oxidative state, as hemoglobin is an antioxidant23, causing cardiomyocytes damage.51 Lack of EPO can directly worsen cardiac function, since it has a direct effect on the contractility of cardiac muscle, prevention of cardiomyocytes apoptosis, stimulation of angiogenesis in the myocardium and improvement endothelial cell function.51 Therefore, anemia contributes to increased cardiac output, development of LVH and HF.48
PRESSURE AND VOLUME OVERLOAD
Chronic kidney disease patients tend to present both pressure and volume overload. The first often results from hypertension and arteriosclerosis, and occasionally from aortic stenosis. The second may be due to arteriovenous fistula – in hemodialysis patients – anemia and hypervolemia.31 Pressure overload results in increased afterload, whereas volume overload results in increased preload.
Both contribute to the development of LVH as an adaptive remodeling process. Increased afterload leads to a concentric thickening of the LV wall (concentric hypertrophy), in order to boost the intraventricular systolic pressure. Volume overload leads to LV dilation (eccentric LVH), by accumulation of new sarcomeres in series. Both patterns of LVH, likewise a mixed pattern, are common in CKD patients, since afterload and preload factors often coexist, having an additive or synergistic outcome.52
Left ventricular hypertrophy development is induced by integrins, which initiate intracellular signaling in response to a stretch of the extracellular matrix. Angiotensin II and endothelin 1 are also thought to stimulate cardiomyocytes intracellular signaling pathways in response to stretch.31 Despite being a beneficial adaptive response at first, myocardial hypertrophy is accompanied by decreased capillary density, causing ischemia.
This promotes cardiomyocytes apoptosis, extracellular matrix and collagen accumulation, interstitial fibrosis and LV stiffening.52 While loss of cardiomyocytes contributes to LV dilation and eventually systolic dysfunction, myocardial fibrosis leads to diminished cardiac compliance and ultimately diastolic dysfunction.31
ALBUMINURIA
Albuminuria is an established biomarker of CKD (Table II). It is also one of the strongest non-traditional cardiovascular risk factors. Mechanisms underlying this association are still unknown.23
In HF patients, there is an increased prevalence of albuminuria.53 Micro- and macroalbuminuria occur in 20-53% and 5-10%, respectively, of HF patients.54 A higher urinary albumin-to-creatinine ratio (UACR) is associated with increased overall cardiovascular mortality and more recurrent hospitalization for HF and specially predicts progression to HFpEF (in comparison to HFrEF).53 UACR is a prognosis marker, associated with increased RV and LV remodeling and longitudinal systolic dysfunction in HFpEF.53
Albuminuria is associated with increased LVM, but possible mechanisms linking this biomarker to changes in the heart have not been identified.53
HYPERHOMOCYSTEINEMIA
The kidney is a major site of homocysteine metabolism. Its accumulation is a significant contributor to the development and progression of kidney disease. Hyperhomocysteinemia is a serious risk factor for CKD and ESRD.55 High levels of homocysteine are also an important risk factor for HF.56
Hyperhomocysteinemia is common in CKD patients and contributes to adverse cardiac remodeling and HF.57 Homocysteine levels are inversely correlated with diastolic function. An association between hyperhomocysteinemia and diastolic dysfunction has also been established.57
OBSTRUCTIVE SLEEP APNEA
Obstructive sleep apnea (OSA) comprises repeated upper airway obstruction, resulting in hypoxemia and sleep fragmentation. Studies have shown an increased incidence of CKD among OSA patients in comparison to controls. On the other hand, renal failure could promote OSA though increased chemoresponsiveness or fluid retention, causing upper airway to collapse in the recumbent position.58
In OSA, repetitive cycles of apneas and hypopneas contribute to greater swings in negative intrathoracic pressure, increasing preload and afterload. This leads to right ventricle distention and consequent limited LV filling, due to diastolic shift of the interventricular septum. OSA also appears to affect cardiac structure and function through increased sympathetic tone, while parasympathetic tone is significantly reduced.59
DIAGNOSIS OF HEART FAILURE IN CHRONIC KIDNEY DISEASE PATIENTS
Heart failure can result from any structural or functional cardiac abnormality, compromising the heart ability to function as a pump that maintains a physiological circulation.52 It is a clinical diagnosis, based on a typical history and physical exam revealing of insufficient cardiac output or cardiac filling to meet systemic demands.20 Diagnosis is often completed with cardiac imaging studies, assessment of fluid status and natriuretic peptides levels.52
In the CKD population, diagnosis of HF can be challenging, contributing to underdiagnosis and undertreatment of this condition.20
CARDIAC IMAGING STUDIES
Echocardiography is an important tool for HF diagnosis, providing accurate measurements for ventricular diameters and volumes, wall thickness, chamber geometry and ejection fraction.3 However, it may be difficult to interpret in CKD patients, as LVH is highly prevalent among this population.20 In the Chronic Renal Insufficiency Cohort study, LVH prevalence in patients without a HF diagnosis and eGFR ≥60,45-59, 30-44 and <30 ml/min/1.73 m2 was 32%, 49%, 57% and 75%, respectively.60 The relation between GFR and LVH remained statistically significant after fully adjusted multivariable models. This emphasizes the prominence of cardiac abnormalities in CKD patients, even in the absence of a HF diagnosis.61 Therefore, structural abnormalities in LVM and geometry may not necessarily aid HF diagnosis among CKD patients.20
ASSESSMENT OF FLUID STATUS
A precise diagnosis of HF is based on signs and symptoms of volume overload (shortness of breath, orthopnea, paroxysmal nocturnal dyspnea, and edema) and signs of increased cardiac filling pressure (jugular venous distension, displaced apical impulse).20 CKD patients may present symptoms and signs of volume overload in the absence of HF, due to systemic volume overload, contributing to a challenging HF diagnosis in this population.20
In hemodialysis patients, assessment of fluid status is done through dry weight evaluation. In most centers, it is clinically determined, based on the lowest body weight a patient can tolerate without developing intraor interdialytic hypotension or other symptoms of dehydration.52 This method lacks specificity, sensitivity and objectivity, so others have been proposed, namely echocardiographic measurement of inferior vena cava diameter and collapsibility, relative plasma monitoring, bioelectrical impedance analysis and echography evaluation of pulmonary congestion.52
B-TYPE NATRIURETIC PEPTIDE (BNP) AND ITS AMINO-TERMINAL FRAGMENT (NT-PROBNP)
Together with atrial natriuretic peptide and C-type natriuretic peptide, BNP is part of the major natriuretic peptides, a group of hormones with an important role in sodium and body volume homeostasis.62
The main stimulus for BNP and NT-proBNP secretion is hemodynamic overload leading to myocardial strecht.7 Consequently to ventricular myocyte stretch, preproBNP is enzymatically cleaved to proBNP and released as BNP (the active hormone form) or NTproBNP (an inactive fragment). Both are released in a1:1 ratio.7
In patients with normal renal function, BNP and NTproBNP are associated with HF severity and LV function, being useful for HF diagnosis, management and prognosis.62 In CKD patients, BNP and NT-proBNP values are influenced by decreased clearance and cardiac damage/stress secondary to renal dysfunction. Reduced eGFR may have an independent inverse correlation with increased BNP levels.7 Therefore, there is a likely need to raise the cut point for HF diagnosis when eGFR is <60 ml/min/1.73 m2.62 In such patients, BNP/NT-proBNP serum concentrations are a weak diagnostic tool and should be carefully interpreted and in relation to GFR, regarding HF diagnosis and assessment of volume status.6
THERAPEUTIC CONSIDERATIONS
The care of patients with coexisting HF and kidney disease is a major medical challenge.63 The majority of CKD-HF patients have HFpEF. For this type of HF, there are no evidence-based recommendations for therapies that improve outcomes (both in the general population and in CKD patients).20 Regarding CKD-HF patients in particular, treatment remains unclear, as there is very little strong evidence to support any recommendations.52
The level of care for HF offered to people with CKD should be the same as is offered to those without CKD. However, medications to improve outcome of HF patients are underused in CKD patients.6
The main goals for HF therapy in CKD consist of the decrease of preload and afterload and reduction of LVH; treatment of myocardial ischemia and inhibition of neurohumoral hyperactivity.52 Therapy goals should contemplate control of fluid volume, treatment of anemia, management of mineral and bone disorders52 and vitamin D deficiency.3 Treatment can include the use of beta-blockers, angiotensin-converting-enzyme inhibitors, angiotensin II receptor blockers, aldosterone antagonists, digitalis glycosides, cardiac resynchronization therapy and optimization of dialysis.52
Decreasing PTH levels substantially attenuates the association between eGFR and LV diastolic dysfunction.64 A small number of studies also showed encouraging changes in LV mass after the correction of anemia, administration of a loop diuretic (for non-dialyzed patients), the implementation of frequent hemodialysis (six times vs three times per week) and kidney transplantation (for dialyzed patients), which appears to point towards the role of volume overload and uremic toxins in LVH.21
In people with CKD and HF, any escalation in therapy and/or clinical deterioration should prompt monitoring of GFR and serum potassium concentration. Special concerns regarding the risk of hyperkalemia should be encouraged by the use of aldosterone antagonists in addition to an angiotensin-converting-enzyme inhibitor or angiotensin II receptor blockers, particularly in patients with lower eGFRs.6
CONCLUSION
Heart failure in CKD has been proposed to be closely related to structural myocardial disease.5 Among CKD patients, abnormalities related to LV structure and function are very common.52 About 40% have LVH, which progressively increases as kidney function declines.6
Alterations in LV structure and geometry likely precede the development of HF.61 Patients with CKD can develop HFrEF, HFmrEF, or HFpEF, the latter more common.20
Most CKD patients develop concentric myocardial hypertrophy with interstitial fibrosis and CKD-associated cardiomyopathy, contributing to left ventricular stiffness, causing diastolic dysfunction leading to HFpEF.6
Heart failure is common among patients with CKD and greatly influences prognosis in this population. Regardless of its prevalence, there are substantial challenges to HF diagnosis and therapy in the CKD population, leading to underdiagnosis and undertreatment.
The pathophysiology underlying HF in CKD patients is not yet well understood, being distinct from the one that occurs in the general population. Indeed, the existence of non-traditional/CKD-specific risk factors for HF in CKD patients has been acknowledged.
A lot remains to be learnt about the physiopathology behind non-traditional/CKD-specific risk factors for HF in CKD patients and its impact in the development of different HF subtypes. Better understanding of such mechanisms could be the key to find appropriate diagnosis and treatment. There is an urgent need for studies in this area, which should take into account the nontraditional risk factors for HF for maximum benefit and proper care for these patients.
Furthermore, the epidemiology of the different HF phenotypes in CKD versus in patients without CKD has not been properly investigated. In both populations, HFpEF is the most prevalent subtype of HF, which we find surprising, given the existence of unique risk factors in CKD patients. Could it be that some of the presumed CKD-specific risk factors play a role in HF development in patients without CKD?
References
1. Beck H, Titze S, Hübner S, Busch M, Schlieper G, Schultheiss U et al. Heart failure in a cohort of patients with chronic kidney disease: the GCKD study. PLoS ONE. 2015;10(4):e0122552. [ Links ]
2. Bright R. Cases and observations illustrative of renal disease accompanied with the secretion of albuminous urine. Guys Hospital Rep. 1836;1:338-79. [ Links ]
3. Liu M, Li X-C, Lu L et al. Cardiovascular disease and its relationship with chronic kidney disease. Eur Rev Med Pharmacol Sci 2014;18(19):2918-26. [ Links ]
4. Bagshaw S, Cruz, D, Aspromonte N et al. Epidemiology of cardio-renal syndromes: workgroup statements from the 7th ADQUI Consensus Conference. Nephrol Dial Transplant. 2010;25(5):1406-16. [ Links ]
5. Chirinos JA, Khan A, Bansal N, et al. Arterial Stiffness, Central Pressures and Incident Hospitalized Heart Failure in the Chronic Renal Insufficiency Cohort (CRIC) Study: Chirinos et al: Large Artery Stiffness, Blood Pressure and Heart Failure in CKD. Circ Heart Fail. 2014;7(5):709-16.
6. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int Suppl. 2013;3:1-150. [ Links ]
7. Nizuma S, Iwanaga Y, Yahata T, Miyazaki S. Renocardiovascular Biomarkers: from the Perspective of Managing Chronic Kidney Disease and Cardiovascular Disease. Front Cardiovasc Med. 2017;6;4:10. [ Links ]
8. Jha V, Garcia-Garcia G, Iseki K et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;20;382(9888):260-72.
9. Lozano R, Naghavi M, Foreman K et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;15;380(9859):2095-128. [ Links ]
10. Rao C, Adair T, Bain C, Doi SA. Mortality from diabetic renal disease: a hidden epidemic. Eur J Public Health 2012;22:280-4. [ Links ]
11. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2016;37:2129-200. [ Links ]
12. Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119(14):e391-479. [ Links ]
13. Maggioni AP, Dahlström U, Filippatos G et al. EURObservational Research Programme: regional differences and 1-year follow-up results of the Heart Failure Pilot Survey (ESCHF Pilot). Eur J Heart Fail. 2013;15(7):808-17. [ Links ]
14. Kemp CD, Conte JV. The pathophysiology of heart failure. Cardiovasc. Pathol 2012;5:365-71. [ Links ]
15. Shah SJ, Kitzman DW, Borlaug BA et al. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation. 2016;134(1):73-90. [ Links ]
16. Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality: a collaborative meta-analysis of general population cohorts. Lancet. 2010;375(9731):2073-81. [ Links ]
17. 2016 USRDS. Annual data report: epidemiology of kidney disease in the United States 2016. [ Links ]
18. Kottgen A, Russell SD, Loehr LR, et al. Reduced kidney function as a risk factor for incident heart failure: the atherosclerosis risk in communities (ARIC) study. J Am Soc Nephrol. 2007;18(4):1307-15. [ Links ]
19. Löfman I, Szummer K, Dahlström U, Jernberg T, Lund LH. Associations with and prognostic impact of chronic kidney disease in heart failure with preserved, mid-range, and reduced ejection fraction. Eur J Heart Fail. 2017;19(12):1606-14. [ Links ]
20. Tuegel C, Bansal N. Heart failure in patients with kidney disease. Heart. 2017;103(23):1848-53. [ Links ]
21. Matsushita K, Ballew SH, Coresh J. Influence of chronic kidney disease on cardiac structure and function. Curr Hypertens Rep. 2015;17(9):581. [ Links ]
22. Kochanek M, Said A, Lerma EV. Mineral metabolism in chronic kidney disease. Dis Mon. 2015;61(10):425-33. [ Links ]
23. Pinheiro da Silva AL, Vaz da Silva MJ. Type 4 cardiorrenal syndrome. Rev Port Cardiol. 2016;35(11):601-16. [ Links ]
24. Parker BD, Schurgers LJ, Branderburg VM, et al. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Ann Intern Med. 2010;152(10):640-8. [ Links ]
25. Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393-408. [ Links ]
26. Scialla JJ, Xie H, Rahman M, et al. Fibroblast factor-23 and cardiovascular events in CKD. J Am Soc Nephrol. 2014;2:349-60. [ Links ]
27. Ix JH, Katz R, Kestenbaum BR, et al. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study), J Am Coll Cardiol 2012;60(3):200-7. [ Links ]
28. Scialla JJ, Lau WL, Reilly MP, et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013;83(6):1159-68. [ Links ]
29. Tanenbaum ND, Quarles LD – Bone diseases in chronic kidney disease – Primer on Kidney Diseases. 5th edition. National Kidney Foundation, 2008:64. [ Links ]
30. Townsend RR. Arterial stiffness and chronic kidney disease: lessons from the Chronic Renal Insufficiency Cohort study. Curr Opin Nephrol Hypertens. 2015;24(1):47-53. [ Links ]
31. Alhaj E, Alhaj N, Rahman I, Niazi TO, Berkowitz R, Klapholz M. Uremic cardiomyopathy: an underdiagnosed disease. Congest Heart Fail. 2013;19(4):e40-5. [ Links ]
32. Chrapko B, Grzebalska A, Nocuń A, Książek A, Drop A. Cardiac sympathetic hyperactivity in chronic kidney disease – a comparison between haemodialysis and peritoneal dialysis patients. Nucl Med Rev Cent East Eur. 2014;17(2):75-82. [ Links ]
33. Levitan D, Massry SG, Romoff M, Campese VM. Plasma catecholamines and autonomic nervous system function in patients with early renal insufficiency and hypertension: effect of clonidine. Nephron. 1984;36(1):24-9. [ Links ]
34. Laederach K, Weidmann P: Plasma and urinary catecholamines as related to renal function in men. Kidney Int. 1987;31(1):107-11. [ Links ]
35. de Beus E, de Jager R, Joles JA, Grassi G, Blankestijn PJ. Sympathetic activation secondary to chronic kidney disease: Therapeutic target for renal denervation? J Hypertens. 2014;32(9):1751-61. [ Links ]
36. Neumann J, Ligtenberg G, Klein II, Koomans HA, Blankestijn PJ. Sympathetic hyperactivity in chronic kidney disease: Pathogenesis, clinical relevance, and treatment. Kidney Int. 2004;65(5):1568-76. [ Links ]
37. Siddiqi L, Prakken NH, Velthuis BK, et al. Sympathetic activity in chronic kidney disease patients is related to left ventricular mass despite antihypertensive treatment. Nephrol Dial Transplant. 2010;25(10):3272-7. [ Links ]
38. Tsuruya K, Eriguchi M, Yamada S, Hirakata H, Kitazono T. Cardiorenal Syndrome in End-Stage Kidney Disease. Blood Purif. 2015;40(4):337-43. [ Links ]
39. Akchurin OM, Kaskel F. Update on inflammation in chronic kidney disease. Blood Purif. 2015;39(1-3):84-92. [ Links ]
40. Colombo PC, Ganda A, Lin J, et al. Inflammatory activation: Cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail Rev. 2012;17(2):177-90. [ Links ]
41. Kerr JD, Holden RM, Morton AR, et al. Associations of epicardial fat with coronary calcification, insulin resistance, inflammation, and fibroblast growth factor-23 in stage 3-5 chronic kidney disease. BMC Nephrol. 2013;14:26. [ Links ]
42. Kalra D, Silvasubramanian N, Mann DL. Angiotensin II induces tumor necrosis factor biosynthesis in the adult mammalian heart through a protein kinase C-dependent pathway. Circulation. 2002:105(18):2198-205. [ Links ]
43. Prabhu S, Chandrasekar B, Murray DR, Freeman GL. Beta-adrenergic blockade in developing heart failure: effects on myocardial inflammatory cytokines, nitric oxide, and remodeling. Circulation. 2000;101(17):2103-9. [ Links ]
44. Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM. TNF-α and IL-1β increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010;47(4):378-86. [ Links ]
45. Zha Y, Qian Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients. 2017;9(3):208. [ Links ]
46. Modaresi A, Nafar M, Sahraei, Z. Oxidative Stress in Chronic Kidney Disease. Iran J Kidney Dis. 2015;9(3):165-79. [ Links ]
47. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301(6):2181-90. [ Links ]
48. Zadrazil J, Horák P. Pathophysiology of anemia in chronic kidney diseases: A review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2015;159(2):197-202. [ Links ]
49. De Lima GA, Mazzali M, Gentil AF, Plotegher L, Grotto HZ. Anemia in chronic renal disease: evaluation of inflammatory activity on eytrhropoiesis and iron metabolism in patients not submitted to dialysis treatment, Clin Lab. 2012;58:695-704. [ Links ]
50. Shah R, Agarwal AK. Anemia associated with chronic heart failure: current concepts. Clin Interv Aging. 2013;8:111-22. [ Links ]
51. Silverberg DS, Wexler D, Iaina A. The role of anemia in the progression of congestive heart failure. Is there a place for erythropoietin and intravenous iron? J Nephrol. 2004;17(6):749-61. [ Links ]
52. Segall L, Nistor I, Covic A. Heart failure in patients with chronic kidney disease: a systematic integrative review. Biomed Res Int. 2014;2014:937398. [ Links ]
53. Katz DH, Burns JA, Aguilar FG, Beussink L, Shah SJ. Albuminuria is independently associated with cardiac remodeling, abnormal right and left ventricular function, and worse outcomes in heart failure with preserved ejection fraction. JACC Heart Fail. 2014;2(6):586-96. [ Links ]
54. Metra M, Cotter G, Gheorghiade M, Dei Cas L, Voors AA. The role of the kidney in heart failure. Eur Heart J. 2012;33(17):2135-42. [ Links ]
55. Ostrakhovitch EA, Tabibzadeh S. Homocysteine in Chronic Kidney Disease. Adv Clin Chem. 2015;72:77-106. [ Links ]
56. Strauss E, Supinski W, Radziemski A, Oszkinis G, Pawlak AL, Gluszek J. Is hyperhomocysteinemia a causal factor for HF? The impact of the functional variants of MTHFR and PON1 on ischemic and non-ischemic etiology. Int J Cardiol. 2017;228:37-44. [ Links ]
57. Rafeq Z, Roh JD, Guarino P, Kaufman J, Joseph J. Adverse myocardial effects of B-vitamin therapy in subjects with chronic kidney disease and hyperhomocysteinaemia. Nutr Metab Cardiovasc Dis. 2013;23(9):836-42. [ Links ]
58. Marrone O, Battaglia S, Steiropoulos P, et al. Chronic kidney disease in European patients with obstructive sleep apnea: the ESADA cohort study. J Sleep Res. 2016;25(6):739-45. [ Links ]
59. Triposkiadis F, Giamouzis G, Parissis J, et al. Reframing the association and significance of co-morbidities in heart failure. Eur J Heart Fail. 2016l;18(7):744-58.
60. Park M, Shlipak MG, Katz R, et al. Subclinical cardiac abnormalities and kidney function decline: the multi-ethnic study of atherosclerosis. Clin J Am Soc Nephrol. 2012;7(7):1137-44.
61. Denker M, Boyle S, Anderson AH, et al. Chronic Renal Insufficiency Cohort Study (CRIC): Overview and Summary of Selected Findings. Clin J Am Soc Nephrol. 2015;10(11):2073-83. [ Links ]
62. Iwanaga Y, Miyazaki S. Heart Failure, chronic Kidney Disease, and biomarkers – an integrated viewpoint. Circ J. 2010;74(7):1274-82. [ Links ]
63. Schefold JC, Filippatos G, Hasenfuss G, Anker SD, von Haehling S. Heart failure and kidney dysfunction: epidemiology, mechanisms and management. Nat Rev Nephrol. 2016;12(10):610-23. [ Links ]
64. Park M, Hsu CY, Li Y, et al. Associations between kidney function and subclinical cardiac abnormalities in CKD. J Am Soc Nephrol. 2012;23(10):1725-34. [ Links ]
Luísa Viveiros, MD
Faculty of Medicine of Porto University
Alameda Prof. Hernâni Monteiro, 4200-319 Porto, Portugal
E-mail: mimed12232@med.up.pt
Disclosure of potential conflicts of interest: None declared.
Received for publication: Mar 30, 2018
Accepted in revised form: Nov 16, 2018