










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.3 Lisboa set. 2018
CASE REPORT
Light chain deposition disease: atypical associations in a rare disease
Ivo Cunha1, Sandra Silva2, Rui Henrique3, Maximino Costa2
1 Serviço de Medicina Interna do Hospital Pedro Hispano
2 Unidade de Nefrologia do Hospital Pedro Hispano
3 Serviço de Anatomia Patológica do IPO Porto
ABSTRACT
Light chain deposition disease is a systemic disorder characterized by deposition of monoclonal light chains in various organs. We present a case of a 58#8209;year#8209;old woman who was referred for a nephrology consultation due to worsening renal function and nephrotic range proteinuria. The diagnosis work#8209;up mentioned a plasmatic creatinine of 1.5mg/dL (estimated glomerular filtration rate of 38 mL/min/1.73m2 by MDRD equation), a urinary sediment with microhematuria, a protein/creatinine ratio of 5, a seric and urinary immunoelectrophoresis compatible with a monoclonal gammopathy IgG/Kappa, hypocomplementemia, type II cryoglobulinemia and a pulmonary nodule of irregular shape 15mm in diameter. A renal biopsy was performed and showed a marked expansion of the mesangium with nodules of amorphous material and small outbreaks of tubular atrophy associated with interstitial fibrosis. The nodules were PAS positive and Congo red negative and stained for light chain kappa by immunocytochemistry. Immunofluorescence was negative for IgA, IgG, IgM, C1q and C3c. A diagnosis of light chain deposition disease was made and concomitant multiple myeloma excluded. Treatment was initiated with bortezomib, dexamethasone and thalidomide with complete hematological remission and improvement in renal function. She also showed normalization of the cryoglobulinemia and disappearance of the pulmonary nodule previously detected, despite worsening of cardiac function as a result of the chemotherapy implemented. This clinical case highlights the well‑known renal involvement in light chain deposition disease, but also some atypical clinical associations, namely type II cryoglobulinemia and pulmonary nodule disease.
Keywords: Cryoglobulinemia, light chain deposition disease, monoclonal immunoglobulin deposition disease, pulmonary nodule.
INTRODUCTION
Monoclonal immunoglobulin deposition disease (MIDD) is a group of multisystem disorders characterized by light or heavy chains deposition of monoclonal immunoglobulins in various organs1,2. The most commonly diagnosed MIDD is light chain deposition disease (LCDD), followed by heavy chain deposition disease and light and heavy chain deposition disease. LCDD is a rare disease with a male predominance and a median age at diagnosis of 58 years old. It is usually associated with monoclonal gammopathies of undetermined significance in 17% and with multiple myeloma (MM) in 58% of patients2,4#8209;5.
In LCDD there is a single clone of plasma cells responsible for the overproduction of kappa, in the majority of cases, or sometimes lambda light chains3. Clinical manifestations vary, depending on which organs are predominantly involved, with the kidney one of the most commonly affected. This is explained by the fact that light chains (LCs) are filtered by the glomeruli, reabsorbed in proximal tubules by receptor mediated endocytosis, and degraded in tubular cells by lysosomal enzymes. The clinical scenario might be one of a glomerulopathy, including rapidly progressive glomerulonephritis or an acute tubulointerstitial nephritis1,4 revealed by renal lesion, hypertension, microhematuria, proteinuria and more rarely renal tubular acidosis. The characteristic pathologic features of renal LCDD include nodular sclerosing glomerulopathy by light microscopy; diffuse linear staining of glomerular and tubular basement membranes for a single light chain by immunofluorescence; and nonfibrillar, powdery electron‑dense deposits in basal membranes detected by electron microscopy6. Symptomatic extrarenal deposition is rare2 but hepatic, cardiac, pulmonary14 and neural deposits have been documented.
The clinical course of an untreated disease is one of inexorably progressive chronic kidney disease (CKD) and death. In light of the scarcity of published studies, the treatment is based in chemoterapy and autologous stem cell transplant1,5#8209;10 in selected cases.
CASE REPORT
A 58#8209;year#8209;old Caucasian woman with an history of hypertension controlled with an ACEI, a thiazide and a beta‑blocker with no clinically known micro or macrovascular involvement, active tabagism with chronic bronchitis and a mitral valve rheumatic stenosis was referred for a nephrology consultation due to worsening renal function with a plasmatic creatinine (pCr) of 1.3mg/dL (estimated glomerular filtration rate (GFR) of 42mL/min/1.73m2 by MDRD‑4 equation) and nephrotic range proteinuria (3537mg in a 24h urine specimen). The work#8209;up showed a serum creatinine of 1.5mg/dL (estimated GFR of 38 mL/min/1.73m2 by MDRD‑4 equation), a urinary sediment with microhematuria, a protein/creatinine ratio of 5, a protein determination of 4.2g in a 24h urine specimen (with 72% albumin), a serum albumin of 3.3 g/dL, and a total protein of 5.2g/dL. A kidney ultrasound showed normal#8209;sized kidneys with normal parenchyma and no signs of lithiasis or obstructive uropathy. A seric electrophoresis showed a gamma spike; seric and urinary immunoelectrophoresis were compatible with a monoclonal gammopathy IgG/Kappa, and the serum kappa/lambda ratio was 1.53. Serum free light chains determination was not available in our institution.
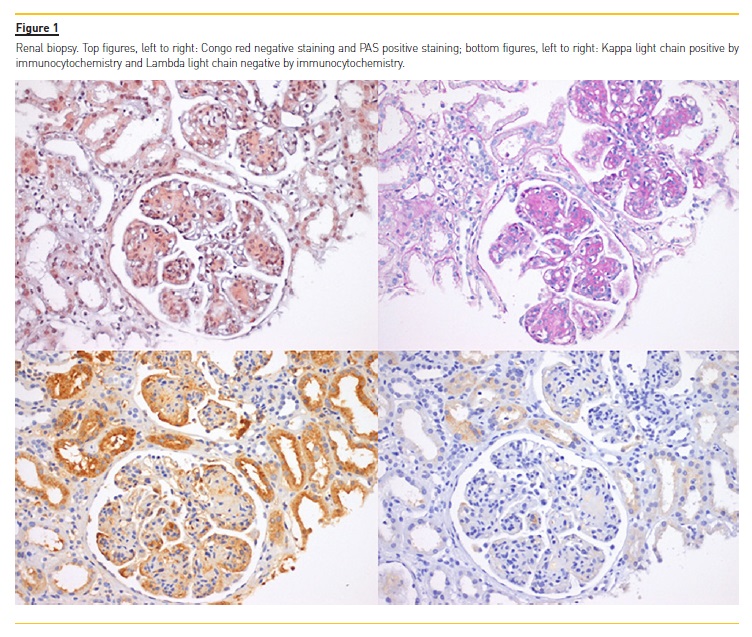
She had normal liver‑function tests, an unremarkable complete blood count, and unremarkable peripheral blood immunophenotyping by flow cytometry. The serological work#8209;up showed normal immunoglobulin levels, hypocomplementemia (C3 of 63mg/dL (N 83‑193) and C4 of 13.3mg/dL (N 15‑57)) and a type II cryoglobulinemia (82mcg/mL (N<20); IgG with polyclonal features and a monoclonal fraction IgG/Kappa without rheumatoid activity). ANCAs, ANAs, ENAs, rheumatoid factor, anti‑dsDNA, anti‑GBM were negative and she also had negative serology for HIV and HCV (including cryocrit negative for HCV antibody). A prior HBV infection was detected (anti‑HBs and anti#8209;HBc positives and HBsAg negative). Viral load was not measured. A transthoracic echocardiography showed preserved systolic function, normal sized chambers without hypertrophy apart from the already documented moderate mitral insufficiency; electromyography study was also normal. A pulmonary nodule of irregular shape 15mm in diameter, not present one year prior in a similar exam, was observed in a thorax CT. A renal biopsy was performed (Figure 1): glomeruli showed a marked expansion of the mesangium frequently with nodules of amorphous material; small outbreaks of tubular atrophy and interstitial fibrosis were also present. Glomerular nodules were PAS positive, Congo red negative and displayed restriction for kappa light chain by immunocytochemistry, and were negative for lambda light chain. Immunofluorescence microscopy was negative for IgA, IgG, IgM, C1q and C3c. A diagnosis of LCDD was performed with concomitant exclusion of an MM by bone marrow aspirate and biopsy with immunophenotyping showing <1% plasmocytes, normal cellularity and no identifiable bony or extramedullary plasmacytoma by means of computed tomography (CT). Treatment was started with bortezomib, dexamethasone and thalidomide for six cycles with complete hematological remission, resolution of the cryoglobulinemia and hypocomplementemia and disappearance of the pulmonary nodule. Despite that, there was progression to CKD stage 4/A1. Due to chemotherapy there was also worsening of cardiac function with development of a dilated cardiomyopathy with global hypokinesis and a marked ventricular function depression of 28% of non‑ischemic etiology as suggested by the cardiac MRI and also a peripheral neuropathy in relation with thalidomide use.
DISCUSSION
In the case presented, the clinical scenario was predominantly a glomerulopathy, with hematuria and a nephrotic range proteinuria. With the results of the serum and urine immunoelectrophoresis and the predominance of kappa light chains, the diagnosis of an LCDD was the most likely, and was later histologically confirmed. Due to the high co#8209;association with MM2,4‑5 this pathology was excluded. Our case is exceptional for two particularities – the association with cryoglobulinemia type II and with pulmonary nodular disease.
Regarding the cryoglobulinemia, two clinical scenarios can be postulated: 1) the patient could have had an essential mixed cryoglobulinemia of unknown etiology that could have been clinically silent12,13 until the development of the LCDD; or 2) it could be an unusual manifestation of the LCDD. The last is the most probable cause, as the histological pattern was not one of a renal manifestation of cryoglobulinemia, and mainly because with chemotherapy, there was resolution of the cryoglobulinemia. Pulmonary nodular disease is a described, but still unusual, association with LCDD14.
LCDD rarely involves the respiratory system, mainly the pulmonary parenchyma and the bronchi, and sometimes the interstititium, in the form of a nodular or diffuse pattern14. In our case the nodule formation was already extensively studied in a pulmonology consultation, the previous year. The resolution of the pulmonary image with the treatment of the LCDD suggests a possible, but still unproven due to the lack of a nodular biopsy, association with the LCDD. In relation to the treatment option, a bortezomib‑based scheme is possibly the choice with the best outcomes to achieve a complete (CR) or very good partial response (VGPR)1,2.
Aggressive chemotherapy should be attemped even in patients with advanced renal diseases as it has been demonstrated that an improvement in renal function could be achieved by the end of the first year if a clonal response can be sustained1. Also, following the same studies, its unquestionable that after achievement of a CR/VGPR, the next step is autologous stem cell transplant (ASCT)1,15‑16.
While the best choice for a chemotherapy scheme, proteasome inhibitors can induce cardiotoxicity17. In our case the echocardiography changes were concomitant with the chemotherapy with confirmation on the cardiac MRI, which showed a dilated cardiomyopathy with a global hypokinesis and marked ventricular function depression, the typical pattern in cases of chemotherapy induced cardiotoxicity.
Thalidomide could also have been an adjuvant, but is typically associated with sinus node dysfunction, bradyarrhythmias and heart block17. Even so, the incidence of heart failure under bortezomib is relatively low (up to 4%), with meta‑analyses and other investigations18 showing conflicting results with regards to this risk, apart from that it could be exacerbated by the concomitant use of steroids19,20. In the absence of a known cardiac condition with clinical heart failure, as was the case, it is the best choice for chemotherapy. Proving this association is the slight improvement in the cardiac function in the latest transthoracic echocardiography.
Cardiac dysfunction is the reason for not performing ASCT at this time. Peripheral neuropathy that could have been induced by both thalidomide and bortezomib, as it also improved at the end of treatment.
In conclusion, LCDD is a systemic disease with a poor outcome, if not detected and treated aggressively early in its course. The differential diagnosis is vast, as its clinical manifestations and associations can be, with kidney biopsy playing a vital role in its differentiation.
References
1. Sayed RH, et al. Natural history and outcome of light chain deposition disease. Blood. 2015;126(26):2805–2810. [ Links ]
2. Jimenez‑Zepeda VH, et al. Light chain deposition disease: novel biological insights and treatment advances. International Journal of Laboratory Hematology. 2012;34:47–355. [ Links ]
3. Sanders PW, Herrera GA. Monoclonal immunoglobulin light chain‑related renal diseases. Seminars in Nephrology. 1993;13:324–341. [ Links ]
4. Pozzi C, et al. Light chain deposition disease with renal involvement: clinical characteristics and prognostic factors. American Journal of Kidney Diseases. 2003;42:1154–1163. [ Links ]
5. Pozzi C, Locatelli F. Kidney and liver involvement in monoclonal light chain disorders. Seminars in Nephrology. 2002;22:319–330. [ Links ]
6. Lin J, Markowitz GS, Valeri AM, et al. Renal monoclonal immunoglobulin deposition disease: the disease spectrum. J Am Soc Nephrol. 2001;12(7):1482–1492. [ Links ]
7. Nasr SH, et al. Renal monoclonal immunoglobulin deposition disease:a report of 64 patients from a single institution. Clin J Am Soc Nephrol. 2012;7(2):231–239. [ Links ]
8. Pozzi C, et al. Renal disease and patient survival in light chain deposition disease. Clin Nephrol. 1995;43(5):281–287. [ Links ]
9. Leung N, et al. Long‑term outcome of renal transplantation in light‑chain deposition disease. Am J Kidney Dis. 2004;43(1):147–153. [ Links ]
10. Short AK, et al. Recurrence of light chain nephropathy in a renal allograft. A case report and review of the literature. Am J Nephrol. 2001;21(3):237–240. [ Links ]
11. Hehe EC, et al. Kidney disease associated with plasma cell dyscrasias. Blood. 2010; 2;116(9):1397–1404.
12. Lamprecht P,et al: Mixed cryoglobulinaemia, glomerulonephritis, and ANCA: essential cryoglobulinaemic vasculitis or ANCA‑associated vasculitis? Nephrol Dial Transplant. 1998; 13: 213–221. [ Links ]
13. Ferri C. Mixed cryoglobulinemia. Orphanet J Rare Dis. 2008;3:25. [ Links ]
14. Colombat M, et al. Light chain deposition disease involving the airways: diagnosis by fibreoptic bronchoscopy. Eur Respir J. 2007;29(5):1057–1060. [ Links ]
15. Royer B, et al. High dose chemoterapy in light chain or light and heavy chain deposition disease. Kidney International. 2004;65(220):642–648. [ Links ]
16. Lorenz EC, et al. Long‑term outcome of autologous stem cell transplantation in light chain deposition disease. Nephrol Dial Transplant. 2008;23:2052–2057. [ Links ]
17. Zamorano JL, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines. European Heart Journal. 2016;37:2768–2801. [ Links ]
18. Stewart AK, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142–152. [ Links ]
19. Cole DC, et al. Cardiovascular Complications of Proteasome Inhibitors Used in Multiple Myeloma. Cardiology in Review. 2018;26(3):122–129. [ Links ]
20. Meseeha MG, et al. Partially reversible bortezomib‑induced cardiotoxicity: an unusual cause of acute cardiomyopathy. J Community Hosp Intern Med Perspect. 2015;5:28982. [ Links ]
Ivo Cunha, MD
Serviço de Medicina Interna do Hospital Pedro Hispano
Matosinhos, Portugal
E‑mail: ibooo1@hotmail.com
Disclosure of potential conflicts of interest: none declared.
Received for publication: Jul 03, 2018
Accepted in revised form: Sep 09, 2018


{kind=link}