









Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.3 Lisboa Sept. 2018
CASE REPORT
Immunoglobulin G4-related disease mimicking multiple myeloma
Marta Sofia Costa, Andreia Silva, Luísa Costa, Ana Rodrigues, Tiago Barra, Sérgio Lemos, Jesús Garrido
Department of Nephrology, Centro Hospitalar Tondela-Viseu, Viseu, Portugal
ABSTRACT
Immunoglobulin G4‑related disease (IgG4‑RD) is a rare, poorly understood immune mediated disorder. It is characterized by a wide clinical spectrum depending on the organs affected. Serum IgG4 may be elevated, but this is not mandatory. Imaging abnormalities are usually detected in the affected organs, which typically show enlarged dimensions. Definitive diagnosis is made upon tissue biopsy demonstrating lymphoplasmacytic infiltration with predominance of polyclonal IgG4‑positive plasma cells, storiform fibrosis and obliterative phlebitis. The most common renal manifestations [(IgG4‑related kidney disease (IgG4#8209;RKD)] include tubulointerstitial nephritis, membranous glomerulonephritis and pyelitis. There is, usually, good therapeutic response to corticosteroids, but rituximab may be needed in cases of relapsing or resistant disease. A diagnostic challenge, and because it diagnosis needs specific immunohistochemical staining techniques, IgG4‑RKD should be contemplated in the differential diagnosis of obscure kidney disease in order not to be missed.
Keywords: Immunoglobulin G4; immunoglobulin G4‑related disease; immunoglobulin G4‑related kidney disease; lymphoplasmacytic infiltration; storiform fibrosis; tubulointerstitial nephritis.
INTRODUCTION
Immunoglobulin G4‑related disease (IgG4‑RD) is a multi#8209;organ immune mediated disorder that mimics malignant, infectious and inflammatory conditions.1 It is characterized by lymphoplasmacytic tissue infiltration with the predominance of IgG4‑positive plasma cells and by the development of fibrosis.
Its incidence and prevalence are poorly understood since it was not recognized as an individual pathological entity until 2003.2 One cross#8209;sectional study estimated an incidence of 60:1,000,000 and a prevalence of 8,000/year.3
Unlike the majority of autoimmune disorders, IgG4‑RD has a male predominance4, with a male to female ratio estimated at 2.8:1.5 The mean age at diagnosis is 61.4 years.6
It is thought that this disorder has an allergic background with an anomalous immune response where type 2 T helper cells and regulatory T cells are up regulated.7 This incites an inflammatory cascade that culminates with the differentiation of B cells in IgG4 producing plasma cells.8
Their expansion leads to infiltration of the organs and their dysfunction. Regulatory T cells are also responsible for activating fibroblasts9 and the differentiation of endothelial and epithelial cells into myofibroblasts,10 resulting in tissue fibrosis. No specific target antigen has yet been identified, and the role of IgG4 antibodies remains obscure.
This newly recognised condition links many autoimmune entities once regarded as isolated (Table I).
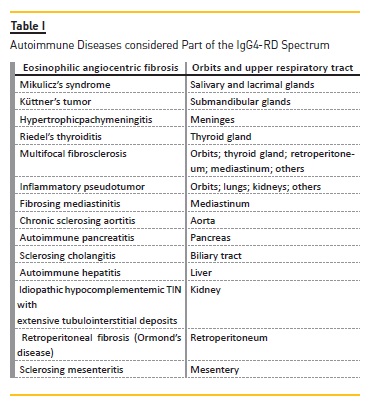
The presentation of IgG4‑RD is usually subacute and the overall signs and symptoms are dictated by the organs involved.
The gold standard for the diagnosis is tissue biopsy demonstrating the characteristic histopathological findings and immunohistochemical staining: lymphoplasmacytic tissue infiltration with predominance of polyclonal IgG4‑positive
plasma cells (30 to 50 IgG4‑positive cells per high power field in most tissues; 10 IgG4‑positive cells per high power field in the kidney) and CD4+ T lymphocytes, fibrosis and obliterative phlebitis.
The fibrosis in IgG4‑RD has a storiform pattern, represented by a cartwheel appearance of the arranged fibroblasts and inflammatory cells.11
IgG4#8209;related kidney disease (IgG4‑RKD) may present as tubulointerstitial nephritis, membranous glomerulonephritis and pyelitis.12
A good therapeutic response to glucocorticoids is characteristic, particularly if excessive tissue fibrosis has not supervened.13 In the presence of a corticoresistant disorder, B cell depletion with rituximab is known to induce clinical response in some cases.14
CASE REPORT
An 80–year–old man, previously referred to the Hematology department for thrombocytopenia, was assumed to have multiple myeloma (MM) IgG kappa and lambda. He had elevated serum levels of IgG [5838 mg/dL (650 – 1600 mg/dL)], kappa [5230 mg/dL (598 – 1329 mg/dL)] and lambda [3630 mg/dL (280 – 665 mg/dL)] light chains and beta‑2‑microglobulin [14,5 mg/L (0.8 – 2.2 mg/L)].
Serum protein electrophoresis showed hypergammaglobulinemia (Figure 1) and kappa and lambda monoclonal bands were detected in serum immunofixation electrophoresis (but not in urine). Bone marrow aspiration and biopsy were inconclusive. Renal function was normal.
After treatment with prednisolone, cyclophosphamide and talidomid he was considered to be in remission.
The patient was referred to the Nephrology department for elevation in serum creatinine levels three years later. Serum creatinine was 1.8 mg/dL (0.4 – 1.2 mg/dL) with a proteinuria of 510 mg in a 24‑hour urine collection and microscopic hematuria in a spot urine sample (46/uL). He had a polyclonal gammopathy with elevation of total serum IgG (4803 mg/dL), kappa and lambda light chains. Serum free light chains were marginally elevated, with a kappa/lambda ratio of 0.92, and no monoclonal bands were detected in serum or urine immunofixation electrophoresis. Specific IgG subclass measurement showed elevation of IgG1 [36200 mg/L (3150 – 8550 mg/L)] and IgG3 [11300 mg/L (230 – 1960 mg/L)], with a mild rise of IgG4 [1650 mg/L (110 – 1570 mg/L)] and normal IgG2. Serum C4 was markedly reduced [1.3 mg/L (12.0 – 36.0 mg/dL)]. The autoimmunity panel revealed positive antinuclear antibodies (ANA) in a 1/1280 titer with a homogeneous pattern and rheumatoid factor [55.7 UI/mL (< 20.0 UI/mL]. Virus markers were negative for hepatitis B and C and HIV.
He had normal sized kidneys on renal ultrasound, with adequate corticomedullary differentiation.
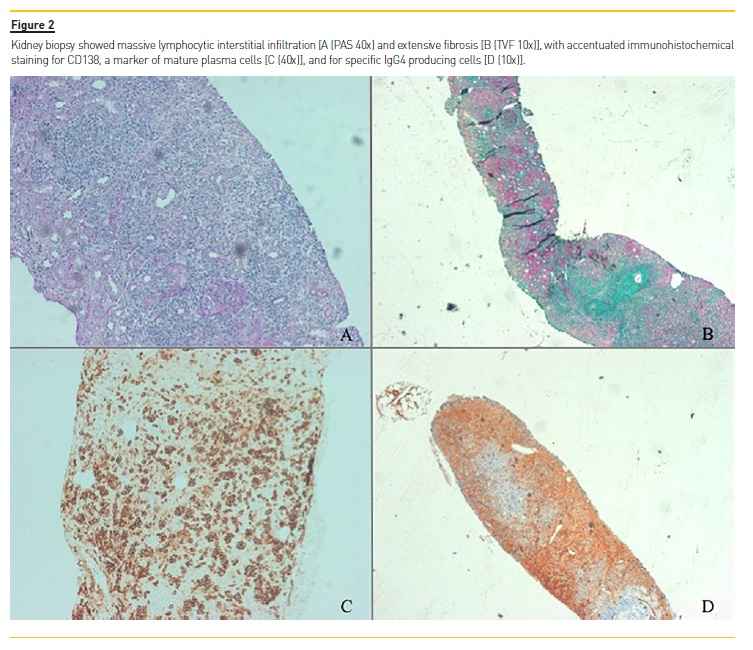
Bone marrow aspiration and biopsy revealed 0.6% of mature plasma cells (with polyclonal characteristics) and no signs of neoplastic disease. Kidney biopsy unveiled massive interstitial infiltration of T and B lymphocytes and plasma cells with extensive tubular destruction (70%) due to storiform fibrosis (Figure 2 – A and B). Immunohistochemical staining was diffusely positive for IgG3 and IgG4 (Figure 2 – C and D) with more than 10 IgG4‑ positive plasma cells per high power field, establishing the diagnosis of tubulointerstitial nephritis (TIN) due to IgG4#8209;RKD.
The patient began prednisolone at a dose of 40 mg/day. In 6 months serum creatinine decreased to 1.5 mg/dL and total IgG to 1476 mg/dL. Proteinuria stabilized at 230 mg/day and microscopic hematuria disappeared.
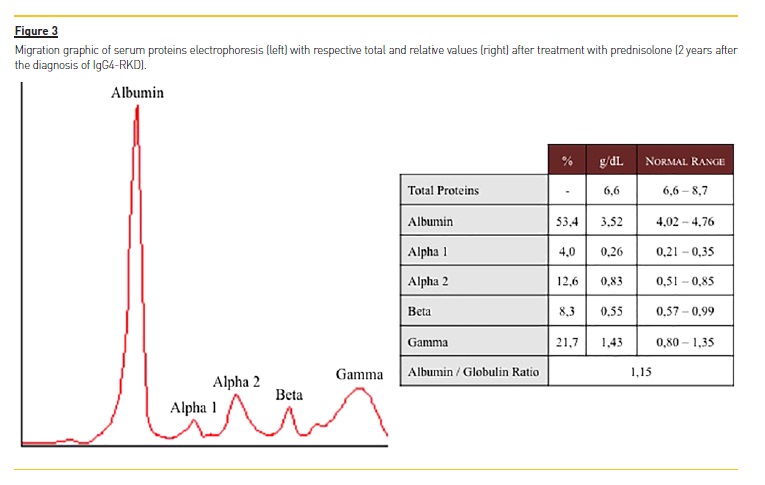
C4 remained markedly low (3.1 mg/dL). Corticotherapy was tapered to a maintenance dose of 10 mg/day and the patient has remained clinically stable since.Figure 3
DISCUSSION
IgG4#8209;RKD has two major clinical presentations: renal dysfunction and imaging oddities. The rise in serum creatinine is usually insidious, but can be rapidly progressive.15 TIN is the most common renal manifestation of IgG4‑RD. Proteinuria and hematuria may be present, but are not usually detected unless there are concomitant glomerular lesions.16 Nephrotic range proteinuria is rare and its presence is suggestive of membranous glomerulonephritis. Peripheral eosinophilia is detected in about 40% of cases associated with IgG4#8209;RD.17 The most common radiological findings are diffuse bilateral renal enlargement and multiple hypodense lesions.18
Umehara et al. proposed an algorithm for diagnosing IgG4‑RD19 whereas Kawano et al. suggested criteria for specific IgG4‑RKD.20 Tissue biopsy with evidence of the characteristic histopathological pattern is mandatory.
Elevated IgG4 levels is a common serological finding in IgG4‑RKD. The main functions of the IgG subclasses are opsonization and complement activation. IgG4 is the least expressed IgG subclass, accounting for 1 to 4% of IgG levels in normal settings.21 Although an elevated serum concentration is not a diagnostic criterion for IgG4‑RD, it is highly suggestive. Carruthers et al. estimated that the sensitivity and the negative predictive values of elevated serum IgG4 level (> 180 mg/dL) are 90% and 96%, respectively, with a low specificity and positive predictive values.22 Because of their unique structural and functional characteristics, IgG4 antibodies undergo single Fab exchange, resulting in a hybrid antibody with two different Fab domains that crosslink with two different antigens.23 The failure to link two identical antigens results in the inability to form immune complexes and, therefore, to initiate cell immunity and activate the complement system.24 Also, in contrast to the other subclasses, IgG4 has a very low affinity for the binding of Fc receptors and C1q, accounting for its unique anti‑inflammatory properties.
When quantifying the levels of IgG subclasses, prozone effect should always be eliminated through successive dilution methods, which was done in this case.
Despite its low positive predictive value, IgG4 levels are a useful tool in monitoring responsiveness to treatment and in predicting relapses.25
Hypocomplementemia is a distinct feature of IgG4‑RD and it seems to be more frequent if the kidney is involved; however only a low proportion of patients present it.26 IgG1 and IgG3 have the highest affinity for C1q binding and complement activation via the classical pathway, which may account for the striking C4 consumption in these cases, as well in the one here described. Complement levels may be used to appraise disease recurrence in IgG4‑RKD.27
Our patient presented with circulating ANA, rheumatoid factor and polyclonal hypergammaglobulinemia and many patients with IgG4‑RD have been shown to have circulating ANA or rheumatoid factor.
There is still controversy as to whether IgG4‑RD is an autoimmune disease. Its association with hypergammaglobulinemia, autoantibody seropositivity and good responsiveness to corticoid therapy has raised this possibility. Some authors defend that the disorder has an allergic background since it is frequently allied with increased serum levels of IgE and eosinophilia.28
However Mattoo et al.29 showed that plasmablast‑derived antibody clones from patients with IgG4‑RD react with autoantigens in the cytosole of Hep‑2 cells, supporting the role of a disease‑specific autoantibody.
Another possibility is that the clonal expansion of IgG4‑positive plasma cells is an induced anti‑inflammatory response to another underlying immune condition. Some researchers have reported elevated plasmablast accumulations in the blood of patients with active systemic lupus erythematosus and rheumatoid arthritis.30-31
When submitted to chemotherapy, the patient went into remission due to treatment with prednisolone and cyclophosphamide. Glucocorticoids are the first line agent for remission induction in all patients with active disease – prednisone 0.6 mg/Kg/day or 30 – 40 mg/day.32 The initial dose is continued for 2 – 4 weeks and then tapered gradually (5 mg every 1 – 2 weeks) to a maintenance dose (5 – 10 mg/day). Additionally, in a prospective cohort study conducted by Yunyun et al. treatment with glucocorticoids alone versus glucocorticoids plus cyclophosphamide were shown to be equally effective at the initial stage of the disease, but the latter had a lower relapse rate than monotherapy overtime.33
Clinical improvement is rapid and a follow‑up serological assessment should be made within two weeks.34
When first line therapy fails to control the disorder, the use of steroid‑sparing agents, such as rituximab, may be needed.35 However, this and other possible therapeutic agents, such as azathioprine, mycophenolate mofetil, methotrexate, tacrolimus and omalizumab, still require more investigation.36
An international consensus statement has been drawn up by the American College of Rheumatology on the treatment of IgG4‑RD.37
In conclusion, IgG4‑RD is still a poorly understood disorder and its etiology is not yet established. It appears to have characteristics of both allergic and autoimmune disorders. Elevated levels of serum IgG4 are highly suggestive and tissue proven biopsy is the gold standard for diagnosis. IgG4‑RKD usually presents as TIN and treatment response depends on the degree of established fibrosis. Glucocorticoids are the first line treatment and rituximab may be administered in cases of resistant or relapsing disease.
Acknowledgments: The authors would like to thank Dr. Fernanda Carvalho, Dr. Helena Viana and Dr. Mário Gois, from the Laboratory of Renal Morphology in Hospital Curry Cabral.
References
1. Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Annu Rev Pathology 2014; 9: 315-47. [ Links ]
2. Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of autoimmune disease. J Gastroenterol 2003; 38: 982-84. [ Links ]
3. Uchida K, Masamune A, Shimosegawa T, Okazaki K. Prevalence og IgG4-related disease in Japan based on nationwide survey in 2009. Int J Rheumatol 2012; 2012: 358-71. [ Links ]
4. Guma M, Firestein GS. IgG4-related diseases. Best Practice & Research Clinical Rheumatology 2012; 26: 425-38. [ Links ]
5. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012; 366: 539-51. [ Links ]
6. Brito-Zéron P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmunity Reviews 2014; 13: 1203-10. [ Links ]
7. Nirula A, Glaser SM, Kalled SL, et al. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol. 2011; 23 (1): 119-24. [ Links ]
8. Tanaka A, Moryiama M, Nakashima H, et al. Th2 and regulatory immune reactions contribute to IgG4 production and the initiation of Mikulicz disease. Arthritis Rheum 2012; 64(1): 254-63. [ Links ]
9. Moriyama M, Tanaka A, Maehara CS, et al. T helper subsets in Sjogrens syndrome and IgG4-related dacryoadenitis and sialoadenitis: a critical review. J Autoimmu 2014; 51: 81-8. [ Links ]
10. Rosenbloom J, Mendoza FA, Jimenez SA. Strategies for anti-fibrotic therapies. Biochim Biophys Acta 2013; 1832 (7): 1088-103. [ Links ]
11. Smyrk TC. Pathological features of IgG4-related sclerosing disease. Curr Opin Rheumatol 2011; 23: 74. [ Links ]
12. Saeki T, Kawano M. IgG4-related kidney disease. Kidney Int. 2014; 85(2): 251-7. [ Links ]
13. Obazaki K, Chiba T. Autoimmune related pancreatitis. Gut 2002; 51: 1. [ Links ]
14. Khosroshahi A, Bloch DB, Deshpande V, et al. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010; 62(6): 1755-62. [ Links ]
15. Cortazar FB, Stone JH. IgG4-related disease and the kidney. Nat Rev Nephrol 2015; 11(10):599-609. [ Links ]
16. Kawano M, Takako S. IgG4-related Kidney disease – an update. Curr Opin Nephrol Hypertens 2015; 24(2): 193-201. [ Links ]
17. Baker RJ, Pusey CD. The changing profile of acute tubulointerstitial nephritis. Nephrol Dial Transplant 2004; 19: 8-11. [ Links ]
18. Cornell LD, et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol 2007; 31:1586-97. [ Links ]
19. Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for the diagnosis of IgG4-related disease. Mod Rheumatol 2012; 22: 21-30. [ Links ]
20. Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol 2011; 15(5): 61-26. [ Links ]
21. Jefferis R, Pound J, Lund J, Goodall M. Effector mechanisms activated by human IgG subclass antibodies: clinical and molecular aspects. Review article. Ann Biol Clin 1994; 52(1): 57-65. [ Links ]
22. Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis 2015; 74: 14-8. [ Links ]
23. Schuurman J, Van Ree R, et al. Normal human immunoglobulin G4 is bispecific: it has two different antigen-combining sites. Immunology 1999; 97(4): 693-8. [ Links ]
24. Kolfschoten M, Schuurman J, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm axchange. Science. 2007; 317 (5844): 1554-7. [ Links ]
25. Tabata T, Kamisawa T, Takuma K, et al. Serial changes of elevated serum IgG4 levels in IgG4-related systemic disease. Intern Med 2012; 50: 69-75. [ Links ]
26. Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 2011; 22: 1343-52. [ Links ]
27. Saeki T, Kawano M, Mizushima I, et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int 2013; 84: 826-33. [ Links ]
28. Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int 2010; 78: 1016-23. [ Links ]
29. Matto H, Mahajan VS, Della-Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol 2014; 134: 679-87. [ Links ]
30. Nicholas MW, Dooley MA, Hogan SL, et al. A novel subset of memory B cells is enriched in autoreactivity and correlates with adverse outcomes in SLE. Clin Immunol 2008; 126: 189-201. [ Links ]
31. Owczarczyc K, Lal P, Abbas AR, et al. A plasmablast biomarker for nonresponse to antibody therapy to CD20 in rheumatoid arthritis. Sci Transl Med 2001; 3(101): 101ra92. [ Links ]
32. Kamisawa T, Okazaki K, Kawa S, et al. Amendment of the Japanese Consensus Guidelines for Autoimmune Pancreatitis, 2013 III. Treatment and prognosis of autoimmune pancreatitis. J Gastrenterol 2014; 49: 961-70. [ Links ]
33. Yunyun F, Yu C, Panpan Z, et al. Efficacy of cyclophosphamide treatment for immunoglobulin G4-related disease with addition of glucocorticoids. Scientific Reports 2017; 7: 6195. [ Links ]
34. Kamisawa T, Zen Y, Pillai S, Stone J. IgG4-related disease. Lancet 2015; 385: 1460-71 [ Links ]
35. Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine 2012; 91: 57-66. [ Links ]
36. Brito-Zerón P, Kostov B, Bosch X, et al. Therapeutic approach to IgG4-related disease. A systematic review. Medicine 2016; 95(26): e4002. [ Links ]
37. Khosroshahi A, Wallace ZS, Crowe JL, et al. International Consensus Guidance Statement on the Management and Treatment og IgG4-Related Disease. Arthritis Rheum 2015; 67(7): 1688-99. [ Links ]
Marta Sofia Costa, MD
Department of Nephrology
Hospital de São Teotónio
Avenida Rei D. Duarte, 3504-509 Viseu
E-mail: marta.sm.costa@gmail.com
Disclosure of potential conflicts of interest: none declared
Received for publication: Apr 25, 2018
Accepted in revised form: Jul 11, 2018

{kind=link}
{kind=link}
{kind=link}