








Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.32 no.3 Lisboa Sept. 2018
REVIEW ARTICLE
Cellular origin and regulation of kidney fibrosis
Judith Schimpf1,2, Rafael Kramann1
1 RWTH Aachen University, Division of Nephrology, Aachen, Germany
2 Department of Internal Medicine III, Nephrology and Dialysis, Feldkirch Academic Teaching Hospital, Feldkirch, Austria
ABSTRACT
Myofibroblasts take a key position as fibrosis driving, matrix secreting cells in kidney fibrosis and are thought to be important therapeutic targets in chronic kidney disease (CKD). However, their origin and activation pattern have been discussed for many years and are still partly unclear. Recently, Gli1+ cells, which reside in the perivascular niche, have been identified as progenitors of fibrosis-causing myofibroblasts. However, Gli1+ cells only account for about 50% of the myofibroblast population and are predominantly located in the kidney medulla. Nevertheless, the data suggests that Gli1+ cells are an important therapeutic target in kidney fibrosis since genetic ablation of these cells significantly ameliorates kidney fibrosis in rodents. Other potential sources of myofibroblasts in the kidney are circulating bone-marrow derived cells, endothelium and epithelium. The current review will discuss the cellular origin of myofibroblasts and potential mechanisms of myofibroblast activation driving fibrosis and CKD.
Keywords: kidney fibrosis, myofibroblasts, Gli1, Hedgehog.
INTRODUCTION
Kidney fibrosis is the final common pathway in chronic kidney disease (CKD), regardless of the primary kidney disease. While it has been known for decades that the amount of cortical interstitial fibrosis in kidney biopsies correlates better than any other structural change with impaired renal function1, no approved drug or therapy for kidney fibrosis exists to date. Given that nearly 11% of the population in the western world suffers from CKD2 with massively increased mortality and morbidity, it is of the utmost importance to decode the underlying molecular and cellular mechanisms and pathways for the development of new therapeutic options. One of the key characteristics of fibrosis across diverse organs is that after kidney injury, activated fibroblasts, known as myofibroblasts, start expanding and produce extracellular matrix (ECM). However, if the activation continues, the process, initially intended to contribute to repair, leads to destruction of kidney tissue and ultimately to irreversible loss of kidney function – End-Stage Renal Disease (ESRD).
In this review we will discuss the debated cellular origin of myofibroblasts and which signaling pathways contribute to their activation and in the end to scar formation in the kidney and functional decline.
C ELLULAR ORIGIN OF MYOFIBROBLASTS
It has been accepted that in various organs, myofibroblasts are the key players in pathologic matrix-production after injury3. Myofibroblasts are of mesenchymal origin and display an extensive rough endoplasmic reticulum, which explains their capability to secrete high amounts of different forms of collagen and other ECM proteins all known to promote fibrosis and express alpha-smooth muscle actin (α-SMA)4. Alpha SMA forms stress fibers, i.e. bundles of myofilaments, that mediate a contractile force and might be involved in scar contraction.
The cellular source of myofibroblasts has been controversial for many years due to the fact that they do not exist in healthy tissues and various experimental evidence points towards different cellular sources (Figure 1). Proximal tubular epithelium, endothelium, circulating cells such as macrophages and fibrocytes and resident mesenchymal cell populations such as pericytes and fibroblasts are among the hotly debated precursor candidates of myofibroblasts. One of the first hypotheses, first formulated about 20 years ago, says that tubular epithelial cells undergo a so-called epithelial-to-mesenchymal transition (EMT) after renal injury5.
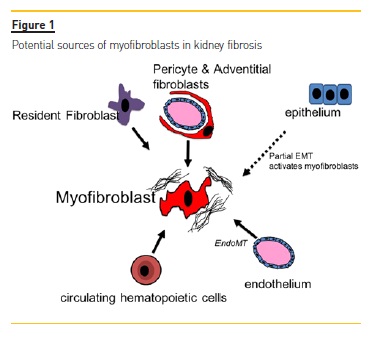
Various genetic fate tracing experiments have reported mixed results, ranging from no influence to major influence on fibrosis6-8. Recent work shows that tubular epithelial cells do not contribute to the direct production of collagen I9 and inducible genetic fate tracing indicates no contribution of proximal tubular epithelium cells to the myofibroblast pool in kidney fibrosis10.
However, tubular epithelium is certainly an important driver of kidney fibrosis and tubular epithelium dedifferentiates in response to injury and acquires many markers of mesenchymal cells in a process that has been recently termed partial EMT11,12. It is now thought that these injured tubular epithelial cells secrete various signals that mediate myofibroblast activation and inflammation. However, the cells do not leave the epithelial basement membrane to become an interstitial myofibroblast10.
The proportion of circulating cells contributing to the myofibroblast pool has been discussed for years13.
In the past, up to one third of the myofibroblast pool was thought to derive from bone marrow-derived mesenchymal stem cells (MSCs)8. Most of these investigations used bone-marrow transplantation techniques, which is tricky since only hematopoietic stem cells engraft whereas other bone marrow cells, such as MSCs, do not engraft well14,15. Bone-marrow derived progenitor cells have been proposed to enter the circulation and home to sites of injury16. However, recent data using state of the art techniques such as inducible genetic fate tracing and single-cell RNA sequencing indicate that only a very small fraction of myofibroblasts derives from circulating cells, i.e. monocytes10.
However, the data also indicates that there is strong cross-talk between resident myofibroblasts and circulating monocytes, with many receptor ligand interaction pairs expressed by both cell-populations. A recent paper by Buchtler et al. suggests that hematopoietic bone marrow derived cells contribute to almost 50% of collagen deposition in the kidney9. Thus the debate on whether hematopoietic cells contribute to collagen deposition and the myofibroblast pool is ongoing and further studies are needed to answer their exact contribution.
Recently, perivascular Gli1+ MSC-like cells were identified as a major cellular origin of kidney fibrosis. Gli1+ cells reside in the perivascular niche across various organs and express the mesenchymal marker platelet derived growth factor receptor beta (PDGFRβ). Using inducible genetic fate tracing in bigenic Gli1CreER;tdTomato mice, we demonstrated that Gli1+ cells expand after injury and transform to fibrosis driving myofibroblasts. Furthermore, ablation of these cells ameliorated fibrosis and restored kidney function15. However, as genetic fate tracing experiments indicate that Gli1+ cells only contribute to a subfraction of myofibroblasts (<50%), further studies are needed to dissect the cellular origin and heterogeneity of all kidney myofibroblasts.
PERICYTES AND CAPILLARY RAREFACTION IN KIDNEY FIBROSIS
The term capillary rarefaction describes the reduction of vascular density along with the functional consequences – hypoxia and impaired hemodynamic and sodium regulatory responses17. Interstitial capillary loss correlates with the degree of interstitial fibrosis18,19.
In progressive kidney disease, peritubular capillaries exhibit significant ultrastructural and functional change independent of the underlying injury20. In diverse experimental sceneries, like unilateral ureteral obstruction21, remnant kidney model22 or experimental glomerulonephritis23 peritubular capillary rarefaction goes along with interstitial fibrosis und tubular atrophy17,20 – which led to the conclusion that regardless of the underlying injury, loss of the peritubular capillary network facilitates fibrosis. However, the mechanisms which connect peritubular capillary loss to kidney fibrosis are still in some part elusive. A longstanding hypothesis suggested that detachment of pericytes from peritubular capillaries drives capillary loss after kidney injury. We were recently able to provide experimental in vivo fate tracing data that indeed supports this hypothesis.
Genetic fate tracing indicated that Gli1+ cells indeed detach from peritubular capillaries after kidney injury and differentiate into myofibroblasts. Furthermore, our data show that genetic ablation of Gli1+ cells in healthy kidneys results in capillary loss, hypoxia and subsequent tubular epithelial injury24. These data suggest that the activation of pericytes and their myofibroblast differentiation initiates a vicious circle that results in fibrosis, capillary loss and tubular damage driving kidney functional decline.
SIGNALING PATHWAYS REGULATING PERICYTE FATE AND ACTIVATION
A key characteristic of fibrosis consists of the reactivation of developmental signaling pathways, which are involved in various processes of kidney fibrosis such as extracellular matrix (ECM) production and myofibroblast differentiation and proliferation, among others. It is beyond the scope of this review to discuss all developmental pathways that have been studied in kidney injury and repair so we will only summarize some recent findings in the Hedgehog, Wnt and Notch signaling pathways.
Since these signaling pathways are activated after injury of the kidney, crosstalk between them seems highly likely. However, probably due to the complexity of their interplay, only a handful of studies which report direct interaction exists25. Better understanding of the interaction between these pathways may facilitate the development of new treatments in kidney fibrosis and CKD. Hedgehog (Hh) signaling has been extensively studied in various cancers such as basal cell carcinoma as well as medulloblastoma and glioma, among others.
Dysregulated Hh signaling has been identified as one of the drivers of cancer progression which lead to the development of smoothened (Smo) antagonists such as vismodegip, which is the first approved canonical Hh inhibitor for advanced basal cell carcinoma26. Recent data, including our own, indicates that Hh signaling is not only a critical driver of cancer progression but also drives fibrotic disease. During canonical Hh signaling, one of the three ligands Indian (Ihh), sonic (shh) or desert (dhh) Hh bind to the receptor patched (ptch1). Upon ligand binding, ptch1 releases its tonic inhibition of the transmembrane protein Smo. Smo then activates the nuclear translocation of the Gli family transcription factor into the nucleus, which results in increased expression of various Hh target genes that partly drive cell-proliferation. Three different Gli proteins, i.e. Gli1, Gli2 and Gli3, have been identified in vertebrates27. While Gli2 responds first to the binding of the Hh ligand, Gli1 serves primarily as an signal amplifier and Gli3 as an repressor28. It has been reported that after kidney injury, tubular epithelial cells (TECs) upregulate the expression of two Hh ligands, namely Ihh and Shh, which result in expression of Hh target genes in myofibroblasts and development of kidney fibrosis29-31.
Using various genetic and pharmacologic models, we have recently shown that Gli2 is an important driver of myofibroblast expansion and fibrosis.32 Furthermore, our work indicates that Hh target genes are also upregulated in human kidney fibrosis32.
Wnt signaling is a highly conserved pathway which plays a central role in diverse biologic processes from embryogenesis to proliferation and carcinogenesis, and there is various evidence that Wnt signaling also drives kidney fibrosis33-35. Similar to Hh signaling, Wnt ligands are expressed by TECs upon injury and drive activation of pericytes and fibrosis36.
Notch signaling describes a cell-cell communication during embryogenesis, which is usually quiescent in adult tissue. However, during fibrosis, Notch signaling is reactivated by binding of the ligands to the Notch receptor family (Notch1-4), leading to a signal cascade which, in the end, results in activation of Notch target genes. Several studies have reported central effects of Notch signaling in kidney fibrosis: Expression of Notch pathway proteins correlates with tubulointerstitial fibrosis and renal function37; human CKD samples showed increased cleaved Notch1 expression, and overexpression of cleaved Notch1 in tubular epithelial cells resulted in kidney fibrosis38. Thus, there are several lines of evidence showing that increased tubular epithelial Notch signaling induces proliferation of interstitial myofibroblasts and drives fibrosis38-43.
CONCLUSION
The cellular origin of kidney fibrosis is still controversial. Most recent data suggest that resident mesenchymal cells such as pericytes are a major source of myofibroblasts in the kidney. The specific contribution of circulating hematopoietic cells to matrix secretion in the kidney is still unsolved but recent single cell RNA-sequencing data suggests that these cells primarily act through indirect mechanisms and activate resident mesenchymal cells. However, further studies are needed to dissect the heterogeneity, cellular origin and mechanism of activation of myofibroblast in mouse and human. The recent development of various single cell genomic tools will certainly help in understanding myofibroblast heterogeneity and crosstalk to other cell types driving CKD progression.
References
1. Schainuck LI, Striker GE, Cutler RE, Benditt EP: Structural-functional correlations in renal disease. II. The correlations. Hum Pathol 1970, 1: 631-641. [ Links ]
2. Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS: Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2003, 41: 1-12. [ Links ]
3. Kramann R, DiRocco DP, Maarouf OH, Humphreys BD: Matrix Producing Cells in Chronic Kidney Disease: Origin, Regulation, and Activation. Curr Pathobiol Rep 2013, 1. [ Links ]
4. Friedman SL, Sheppard D, Duffield JS, Violette S: Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med 2013, 5: 167sr1. [ Links ]
5. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL et al.: Epithelial-tomesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 2015, 21: 998-1009. [ Links ]
6. Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV et al.: Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010, 176: 85-97. [ Links ]
7. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG: Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002, 110: 341-350. [ Links ]
8. LeBleu VS, Taduri G, OConnell J, Teng Y, Cooke VG, Woda C et al.: Origin and function of myofibroblasts in kidney fibrosis. Nat Med 2013, 19: 1047-1053. [ Links ]
9. Buchtler S, Grill A, Hofmarksrichter S, Stockert P, Schiechl-Brachner G, Rodriguez GM et al.: Cellular Origin and Functional Relevance of Collagen I Production in the Kidney. J Am Soc Nephrol 2018. [ Links ]
10. Kramann R, Machado F, Wu H, Kusaba T, Hoeft K, Schneider RK et al.: Parabiosis and single-cell RNA sequencing reveal a limited contribution of monocytes to myofibroblasts in kidney fibrosis. CI Insight 2018, 3. [ Links ]
11. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL et al.: Epithelial-tomesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 2015, 21: 998-1009. [ Links ]
12. Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M et al.: Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med 2015, 21: 989-997. [ Links ]
13. Duffield JS: Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest 014, 124: 2299-2306.
14. Cilloni D, Carlo-Stella C, Falzetti F, Sammarelli G, Regazzi E, Colla S et al.: Limited engraftment capacity of bone marrow-derived mesenchymal cells following T-cell-depleted hematopoietic stem cell transplantation. Blood 2000, 96: 3637-3643. [ Links ]
15. Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA et al.: Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16: 51-66. [ Links ]
16. Kramann R, DiRocco DP, Humphreys BD: Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J Pathol 2013, 231: 273-289. [ Links ]
17. Afsar B, Afsar RE, Dagel T, Kaya E, Erus S, Ortiz A et al.: Capillary rarefaction from the kidney point of view. Clin Kidney J 2018, 11: 295-301. [ Links ]
18. Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T et al.: Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 2012, 82: 172-183. [ Links ]
19. Ishii Y, Sawada T, Kubota K, Fuchinoue S, Teraoka S, Shimizu A: Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int 2005, 67: 321-332. [ Links ]
20. Babickova J, Klinkhammer BM, Buhl EM, Djudjaj S, Hoss M, Heymann F et al.: Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int 2017, 91: 70-85. [ Links ]
21. Ohashi R, Shimizu A, Masuda Y, Kitamura H, Ishizaki M, Sugisaki Y et al.: Peritubular capillary regression during the progression of experimental obstructive nephropathy. J Am Soc Nephrol 2002, 13: 1795-1805. [ Links ]
22. Kang DH, Joly AH, Oh SW, Hugo C, Kerjaschki D, Gordon KL et al.: Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol 2001, 12: 1434-1447. [ Links ]
23. Ohashi R, Kitamura H, Yamanaka N: Peritubular capillary injury during the progression of experimental glomerulonephritis in rats. J Am Soc Nephrol 2000, 11: 47-56. [ Links ]
24. Kramann R, Wongboonsin J, Chang-Panesso M, Machado FG, Humphreys BD: Gli1+ Pericyte Loss Induces Capillary Rarefaction and Proximal Tubular Injury. J Am Soc Nephrol 2017, 28: 776-784. [ Links ]
25. Edeling M, Ragi G, Huang S, Pavenstadt H, Susztak K: Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat Rev Nephrol. 2016, 12: 426-439. [ Links ]
26. Meiss F, Andrlova H, Zeiser R: Vismodegib. Recent Results Cancer Res. 2018, 211: 125-139. [ Links ]
27. Hui CC, Angers S: Gli proteins in development and disease. Annu Rev Cell Dev Biol 2011, 27: 513-537. [ Links ]
28. Kramann R: Hedgehog Gli signalling in kidney fibrosis. Nephrol Dial Transplant 2016, 31:1989-1995. [ Links ]
29. Ding H, Zhou D, Hao S, Zhou L, He W, Nie J et al.: Sonic hedgehog signaling mediates epithelial-mesenchymal communication and promotes renal fibrosis. J Am Soc Nephrol. 2012, 23: 801-813. [ Links ]
30. Fabian SL, Penchev RR, St-Jacques B, Rao AN, Sipila P, West KA et al.: Hedgehog-Gli pathway activation during kidney fibrosis. Am J Pathol. 2012, 180: 1441-1453. [ Links ]
31. Zhou D, Li Y, Zhou L, Tan RJ, Xiao L, Liang M et al.: Sonic hedgehog is a novel tubulederived growth factor for interstitial fibroblasts after kidney injury. J Am Soc Nephrol 2014, 25: 2187-2200. [ Links ]
32. Kramann R, Fleig SV, Schneider RK, Fabian SL, DiRocco DP, Maarouf O et al.: Pharmacological GLI2 inhibition prevents myofibroblast cell-cycle progression and reduces kidney fibrosis. J Clin Invest 2015, 125: 2935-2951. [ Links ]
33. He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y: Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 2009, 20: 765-776. [ Links ]
34. Saito S, Tampe B, Muller GA, Zeisberg M: Primary cilia modulate balance of canonical and non-canonical Wnt signaling responses in the injured kidney. Fibrogenesis Tissue Repair 2015, 8:6. [ Links ]
35. Xiao L, Zhou D, Tan RJ, Fu H, Zhou L, Hou FF et al.: Sustained Activation of Wnt/beta-Catenin Signaling Drives AKI to CKD Progression. J Am Soc Nephrol 2016, 27: 1727-1740. [ Links ]
36. Maarouf OH, Aravamudhan A, Rangarajan D, Kusaba T, Zhang V, Welborn J et al.: Paracrine Wnt1 Drives Interstitial Fibrosis without Inflammation by Tubulointerstitial Cross-Talk. J Am Soc Nephrol 2016, 27: 781-790. [ Links ]
37. Murea M, Park JK, Sharma S, Kato H, Gruenwald A, Niranjan T et al.: Expression of Notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int 2010, 78: 514-522. [ Links ]
38. Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S et al.: Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest 2010, 120: 4040-4054. [ Links ]
39. Djudjaj S, Chatziantoniou C, Raffetseder U, Guerrot D, Dussaule JC, Boor P et al.: Notch-3 receptor activation drives inflammation and fibrosis following tubulointerstitial kidney injury. J Pathol 2012, 228: 286-299. [ Links ]
40. Huang R, Zhou Q, Veeraragoo P, Yu H, Xiao Z: Notch2/Hes-1 pathway plays an important role in renal ischemia and reperfusion injury-associated inflammation and apoptosis and the gamma-secretase inhibitor DAPT has a nephroprotective effect. Ren Fail 2011, 33: 207-216. [ Links ]
41. Sorensen-Zender I, Rong S, Susnik N, Zender S, Pennekamp P, Melk A et al.: Renal tubular Notch signaling triggers a prosenescent state after acute kidney injury. Am J Physiol Renal Physiol 2014, 306: F907-F915. [ Links ]
42. Xiao Z, Zhang J, Peng X, Dong Y, Jia L, Li H et al.: The Notch gamma-secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF-beta/Smad2/3 signaling pathway activation. Int J Biochem Cell Biol 2014, 55: 65-71. [ Links ]
Rafael Kramann, MD
RWTH Aachen University
Division of Nephrology and Clinical Immunology
Pauwelsstrasse 30, 52074 Aachen, Germany
Email: rkramann@ukaachen.de
Disclosure of potential conflicts of interest: none declared.
Received for publication: Sep 09, 2018
Accepted in revised form: Sep 22, 2018