










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.31 no.4 Lisboa dez. 2017
CASE REPORT
Alport Syndrome – A rare presentation
Catarina Neves1, Ana Carolina Cordinhã1, Carmen Ferreira1, Vítor Sousa2,3, Clara Gomes1, António Jorge Correia1
1 Paediatric Nephrology Unit, Hospital Pediátrico de Coimbra, Centro Hospitalar e Universitário de Coimbra, EPE, Coimbra, Portugal
2 Pathology Service, Centro Hospitalar e Universitário de Coimbra, EPE, Coimbra, Portugal
3 Institute of Pathology and Molecular Pathology, Faculdade de Medicina, Universidade de Coimbra, Portugal
ABSTRACT
Introduction: Alport syndrome is a glomerular genetic disease progressing to chronic renal failure associated with deafness and ocular changes. Clinical presentation is usually in the first decade of life with microscopic haematuria and/or persistent proteinuria without hypertension or renal dysfunction. Case Report: Male, 4.5 years old, with an acute nephritic syndrome characterized by macroscopic haematuria, oedema, non-oliguric acute renal failure (maximal urea and creatinine of 25mmol/L and 150μmol/L, respectively), anaemia and proteinuria. Blood pressure normal. Normal immunoglobulins and complement fractions; anti-neutrophil cytoplasmic antibody, anti-nuclear antibody and anti-basal membrane antibody negative. History of recurrent episodes of macroscopic haematuria since the age of eighteen months associated with respiratory infections. No family history of renal disease or deafness. Renal biopsy showed proliferative glomerulonephritis with extracapillary crescentic activity, complete fragmentation of glomerular basement membranes and negative immunofluorescence. Pulse methylprednisolone was given followed by oral prednisolone and cyclophosphamide. Renal function recovered, microscopic haematuria persisted. At age 5, there was reappearance of proteinuria, worsening progressively and two years later he started treatment with enalapril. At age 11, a second renal biopsy revealed mesangial proliferative glomerulonephritis, small foci of glomerular sclerosis and few deposits of IgM on immunofluorescence. He started oral corticosteroids with partial response. Nine months later, bilateral sensorineural deafness was detected. At age 16, he maintains normal renal function, microscopic haematuria with manifest proteinuria. A mutation in homozygosity in the COL4A3 gene, compatible with autosomal recessive Alport syndrome, was identified. Conclusions: This case draws attention to an uncommon early course and clinical/pathological findings of a patient later diagnosed with Alport syndrome, with an initial good response to corticosteroids and cyclophosphamide. The case also illustrates the importance of kidney biopsy, including electron microscopy, in the diagnostic, classification and therapy in kidney diseases with unusual clinical course.
Keywords: Acute renal failure, Alport syndrome, Crescentic glomerulonephritis, Haematuria, Immunosuppression, Proteinuria.
INTRODUCTION
Alport syndrome (AS) is a rare disease of ultrastructural collagen abnormality responsible for 0.2% to 2.3% of all causes of end-stage renal disease (ESRD).1-5 AS is characterized by a progressive form of glomerular disease that is often associated with neural hearing loss and ocular abnormalities.3,4,6-9 The most common transmission is X-linked inheritance (XLAS) (80–85%), arising from mutations in the COL4A5 gene located in Xq22.3.1,3,5-8,10 In autosomal recessive AS (ARAS) (15%) or autosomal dominant AS (ADAS) (5%) mutations of the COL4A3 and COL4A4 genes, respectively, are identified and located in 2q35–37,1,3,5-8,10 and both gender are equally affected.1,4 The prevalence of AS has been estimated at 1:5000–10 000 live-births for XLAS and ARAS 1:50 000 live-births.1,6,11 The median age of onset of ESRD in young untreated patients has been reported to be 22 years.1
Both paediatricians and nephrologists are confronted with difficult situations in which a misdiagnosis has been made in AS patients who previously underwent a renal biopsy and were then treated for other diseases.2 Diagnosis in an adult patient is less difficult than in children because progression has usually occurred, making other diagnoses less likely.2
CASE REPORT
The authors present the case of a four year, eight month old boy who was hospitalized for macroscopic haematuria and oedema. He denied changes in urination, dysuria, increased frequency or urgency. The remaining clinical history and physical examination were normal, including blood pressure. The investigation showed non-oliguric acute renal failure (creatinine 145μmol/L), anaemia (haemoglobin 6.9g/dl), erythrocyturia and proteinuria (urine protein/creatinine ratio (UPCR) 928mg/mmol). Immunoglobulins A, G and M and complement fractions C3 and C4 were normal; anti-neutrophil cytoplasmic antibody, anti-nuclear antibody, anti-basal membrane antibody, B and C hepatitis and HIV serological test results were negative. He had a history of recurrent self-limited episodes of macroscopic haematuria since the age of eighteen months associated with respiratory infections. In these episodes, he had no hypertension, proteinuria or urinary infection. Parents were not consanguineous and there was no family history of renal disease or deafness. The renal biopsy evaluated six glomeruli: two were morphologically normal; two had segmental endocapillary proliferation, with mesangial enlargement and associated hypercellularity; and the two remaining had crescentic extracapillary proliferation, in which epithelial proliferation occupied the entire capillary ball, collapsing the normal structures (Figure 1). In the latter two were small deposits of fibrin and complete fragmentation of glomerular basement membranes with retraction of the capillary ball. At the interstitium level, there was oedema and a slight diffuse inflammatory infiltrate, composed of predominantly lymphocytic cells. There are also slight and focal ischaemic changes of the tubular epithelium, with distalization of proximal tubes and atrophy of the epithelium of the distal tubes with red blood cell casts occupying the lumen (Figure 2). No vascular lesions were observed and immunofluorescence was negative, including for IgA. He was treated with six pulses of methylprednisolone (30mg/kg/dose) followed by oral prednisolone (2mg/kg/day) for four weeks and cyclophosphamide (2mg/kg/day) for twelve weeks. Renal function recovered (creatinine 44μmol/L) and proteinuria and macroscopic haematuria resolved, but microscopic haematuria persisted. A year later there was reappearance of proteinuria (UPCR 89mg/mmol), worsening progressively (UPCR maximum 421mg/mmol), and two years later he started treatment with enalapril 5mg/day. He remained with no macroscopic haematuria, hypertension or renal dysfunction (creatinine 45μmol/L). At age 10, in the context of an upper respiratory infection, he had an episode of macroscopic haematuria, with resolution in three days. At age 11, a second renal biopsy revealed mesangial proliferative glomerulonephritis, small foci of glomerular sclerosis and few deposits of IgM on immunofluorescence.
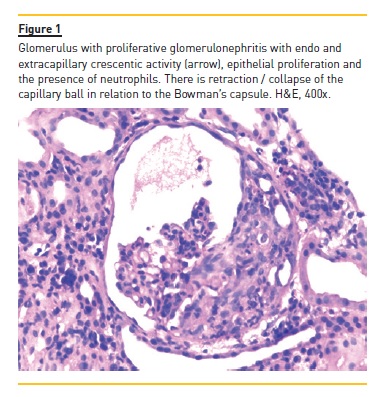
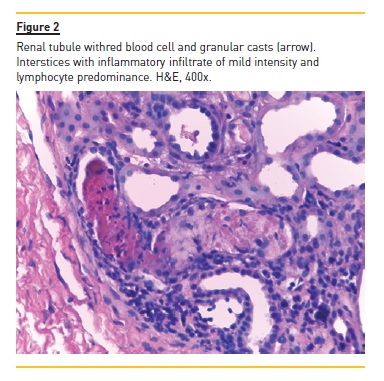
He started oral corticosteroids (prednisolone 2mg/kg/day during eight weeks with subsequent 20mg/48h) and raised the enalapril dose to 20mg/day, with partial response (UPCR 107mg/mmol). Nine months later, bilateral sensorineural deafness was detected, requiring bilateral hearing aids.
Ocular changes and genetic mutation of XLAS were excluded. At age 16 years, he had persistent proteinuria (UPCR 298mg/mmol), microscopic haematuria and normal creatinine (55μmol/L). At this time was identified a mutation c.4441C>T in homozygosity in the COL4A3 gene was identified, which results in a stop codon (p.Arg1481*) in the NC1 domain of the protein, resulting in a truncated protein or in the non-production of the COL4A3 protein. This mutation is compatible with ARAS and he stopped prednisolone.
DISCUSSION
AS was first identified in a British family by Cecil A Alport in 1927, as a dominantly inherited hereditary nephritis characterized by haematuria and nerve deafness.1,2 Phenotypes exhibit a wide spectrum, from mild clinical forms with benign microscopic haematuria, to severe forms evolving to ESRD early in life.2,4,7,9
The fundamental abnormalities lie in the linear and/or the conformational structures of the subchains of collagen IV molecules (COL-IV-α3:α4:α5 or COL-IV-α5:α6:α5), caused by different individual genotypes involving a monogenic mutation in COL4A3, COL4A4 or COL4A5 genes.2-4,6
The presenting renal manifestation is asymptomatic microscopic haematuria, which usually begins in childhood and is manifested by recurrent or persistent haematuria with or without proteinuria, many times precipitated by upper respiratory infections,4,6,10,12-14 as in our case. Increasing proteinuria, hypertension and progressive renal insufficiency occur with time.4,6,10,12-14 ESRD usually presents in males in XLAS and in both sexes in ARAS in the second or third decade of life.6,10,14
ESRD may be the first manifestation.12
Deafness is frequently but not universally present in AS, occurring in approximately 55% of males and 45% of females with the disease, and may be a late and very slowly progressive phenomenon.10 Hearing loss is never congenital, usually becoming apparent by late childhood to early adolescence, and always accompanied by evidence of renal involvement.10 High-frequency sensorineural deafness is the most common hearing loss.12 Disease management entails hearing aids.1 In our case, deafness raised the possibility of this diagnosis, until then not considered.
Ocular defects occur in 15–30% of patients and is bilateral in about 75%.10 In our case, they were excluded.
Anterior lenticonus, corneal posterior polymorphous dystrophy, retinal flecks in the macula and midperipheral fundus are the most common and specific ocular findings of AS.1,12,14 These lesions are absent at birth, usually appearing during the second or third decade of life.10 Megathrombocytopenia is observed occasionally.10,14
ARAS is suggested by the presence of one of the following features: (i) severe early disease in both females and males; (ii) absence of severe signs in parents (they may be completely asymptomatic or may have isolated microscopic haematuria); (iii) parental consanguinity.3,4,8,10 However, clinical and morphologic features are comparable to XLAS.4,5 In our patient we observed severe early renal disease and absence of severe signs in parents.
The diagnosis of AS is usually suspected from the family and/or personal history of renal failure and deafness, as in our case, with or without findings of ocular abnormalities and can subsequently be confirmed or excluded in the majority of cases by the performance of a renal biopsy.4,6 However, family history can be negative.12 Examination of the renal tissue usually excludes other haematuric glomerular disease, the most frequent being IgA nephropathy, and often shows typical ultrastructural abnormalities of the glomerular basement membrane (GBM) when electron microscopy is performed,4 which was not possible in our case.
Light microscopy is widely regarded as of limited value for differentiating between primary glomerulonephritis and AS.2 Diffuse mesangial proliferative changes are observed in most AS patients.2,12 Although rare, the evidence of crescentic proliferation in the pre-transplantation setting of AS has been reported by some authors.6,12,13,15 Nevertheless, it is still unclear if it is an accidental finding, as a super imposition of a morphological characteristic upon a pre-existing case of AS, or a new morphological presentation of this syndrome, possibly associated to a more aggressive clinical course.6,13 A crescentic glomerulonephritis after transplantation, with anti-GBM antibodies, is most often described in the literature.9,11 We can only speculate about the possible pathological mechanisms giving rise to the development of segmental necrosis and cellular crescent formation in patients with AS.15 Normal glomerular intracapillary blood pressure is 40-50mmHg, significantly greater than in other organ capillary beds, which have a pressure of 5-10mmHg.15 Perhaps the combination of the intrinsically high glomerular capillary blood pressure and defective synthesis of collagen IV overwhelms the structural integrity of the GBMs in patients with AS.15 The subsequent rupture of capillary loops could potentially lead to segmental necrosis or cellular crescent formation.15
Early stage AS patients may present as thin basement membrane disease, with or without mesangial lesion.2,4
No significant arterial changes are initially observed.4 Progression of renal lesion from thin GBM disease to focal and segmental glomerular sclerosis has been observed and reported in both XLAS and ARAS patients.2,4,6,12 Re-biopsy usually showed typical changes in electronic microscopic study, as well as more prominent non-specific lesion (including laminated Bowmans capsule and more severe clustering of interstitial foam cells).2,6,12,14 Thin and irregular GBM is the predominant feature in younger children.10,14 In others, the most impressive change is the irregular alternation of thick, thin and normal GBM.10,14 The abnormalities can affect a portion of a capillary loop, an entire loop, some or all loops, or can even spare individual glomeruli.10 In patients with ARAS, GBMs usually show no expression of the α3(IV), α4(IV) or α5(IV) chains, but α5(IV) is expressed in Bowmans capsule, distal tubular basement membranes and epidermal basement membranes.10,14 This pattern of staining may be used to distinguish the XLAS and ARAS.10,14 Immunofluorescence is initially negative; however, faint and irregular deposits of IgG, IgM, and/or C3 may be observed.4
More frequently, granular C3 deposits are irregularly distributed on the glomerular tuft and the afferent arterioles.4 With progression of the lesions, deposits of IgM, C1q, and C3 can be seen in glomerular segmental lesions.4 None of the previous anomalies are pathognomonic.4 However, genetic testing ensure the diagnosis and avoid renal biopsy. On the other hand, a kidney biopsy is usually indicated to characterize the degree and type of kidney involvement, and may be helpful in guiding renoprotective therapy.
There is no specific treatment for AS.6 Angiotensinconverting enzyme inhibitors have been used to retard the progression of the disease, and they are particularly prescribed for those patients with proteinuria.1,6
Another pharmacological therapy is cyclosporine, which can suppress proteinuria and stabilize renal function and histological changes.6 Either dialysis or transplantation can be performed in patients who develop ESRD.1,6
There is no reference in the literature about the use of corticosteroids or cyclophosphamide in AS, but in our case we noticed a significant improvement, with normalization of renal function and resolution of proteinuria and macroscopic haematuria.
CONCLUSION
This case draws attention to uncommon clinical and pathological findings of AS: acute renal failure and cellular crescent formation. In the literature, the presence of crescents in a renal biopsy can be interpreted as a marker of unfavourable outcome and therefore identify patients at a higher risk of rapid renal function deterioration.
In our case, there was a good response to corticosteroids and cyclophosphamide with initial normalization of serum creatinine and proteinuria. It is not clear whether the crescentic nephritis in this case represents a progressive variant of AS or a coincidental superimposed disease. The case also illustrates the importance of a kidney biopsy (and sometimes the importance of repeat kidney biopsies), including electron microscopy in the diagnostic, classification and therapeutic decision-making in genetic kidney diseases with unusual clinical course.
References
1. Ghosh S, Singh M, Sahoo R, Rao S. Alport syndrome: a rare cause of uraemia. BMJ Case Rep. 2014 Feb 13;2014. pii: bcr2013201731. doi: 10.1136/bcr-2013-201731. [ Links ]
2. Yao XD, Chen X, Huang GY, et al. Challenge in pathologic diagnosis of Alport syndrome: evidence from correction of previous misdiagnosis. Orphanet J Rare Dis. 2012 Dec 21;7:100. doi: 10.1186/1750-1172-7-100. [ Links ]
3. Afonso A, Valente I, Macedo L, et al. Alport syndrome – a rare histological presentation. Port J Nephrol Hypert. 2010;24:51-5. [ Links ]
4. Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Am Soc Nephrol. 2009; 20:1210-5. [ Links ]
5. Chang A, Logar CM, Finn LS, et al. A rare cause of necrotizing and crescentic glomerulonephritis in a young adult male. Am J Kidney Dis. 2005;45:956-60. [ Links ]
6. Chugh KS, Sakhuja V, Agarwal A, et al. Hereditary nephritis (Alports syndrome) – clinical profile and inheritance in 28 kindreds. Nephrol Dial Transplant. 1993;8:690-5. [ Links ]
7. Harris JP, Rakowski TA, Argy WP Jr, Schreiner GE. Alport syndrome representing as crescentic glomerulonephritis: a report of two siblings. Clin Nephrol. 1978;10:245-9. [ Links ]
8. Gubler M, Levy M, Broyer M, et al. Alports syndrome. A report of 58 cases and a review of the literature. Am J Med. 1981;70:493-505. [ Links ]
9. Marcocci E, Uliana V, Bruttini M, et al. Autosomal dominant Alport syndrome: molecular analysis of the COL4A4 gene and clinical outcome. Nephrol Dial Transplant. 2009;24:1464-71. [ Links ]
10. Prakash S, Chung KW, Sinha S, et al. Autosomal dominant progressive nephropathy with deafness: linkage to a new locus on chromosome 11q24. J Am Soc Nephrol. 2003;14:1794-803. [ Links ]
11. Mazzucco G, Barsotti P, Muda AO, et al. Ultrastructural and immunohistochemical findings in Alport s syndrome: a study of 108 patients from 97 Italian families with particular emphasis on COL4A5 gene mutation correlations. J Am Soc Nephrol. 1998;9:1023-31. [ Links ]
12. Kashtan CE, Michael AF. Alport syndrome. Kidney Int. 1996;50:1445-63. [ Links ]
13. Gubler MC, Knebelmann B, Beziau A, et al. Autosomal recessive Alport syndrome: immunohistochemical study of type IV collagen chain distribution. Kidney Int. 1995;47:1142-7. [ Links ]
14. Antignac C, Knebelmann B, Drouot L, et al. Deletions in the COL4A5 collagen gene in X-linked Alport syndrome - characterization of the pathological transcripts in nonrenal cells and correlation with disease expression. J Clin Invest. 1994;93:1195-207. [ Links ]
15. Goldman M, Depierreux M, De Pauw L, et al. Failure of two subsequent renal grafts by anti-GBM glomerulonephritis in Alports syndrome: case report and review of the literature. Transpl Int. 1990;3:82-5. [ Links ]
Catarina Neves, MD
Address: Hospital Pediátrico de Coimbra, Centro Hospitalar e Universitário
de Coimbra, EPE, Avenida Afonso Romão-Alto da Baleia,
3000-602 Coimbra, Portugal
Phone: +351960375778
Email: catarinarneves@hotmail.com
Disclosure of potential conflicts of interest: none declared.
Contributors Statements:
– Dr Neves contributed to literature search, analysis and interpretation of the case, drafted the initial manuscript, reviewed and revised the manuscript, and approved the final manuscript as submitted.
– Dr Cordinhã contributed to literature search and analysis and interpretation of the case, reviewed and revised the manuscript, and approved the final manuscript as submitted.
– Dr Ferreira contributed to literature search and analysis and interpretation of the case, reviewed and revised the manuscript, and approved the final manuscript as submitted.
– Dr Sousa contributed to the assessment and images of the renal biopsies, reviewed and revised the manuscript, and approved the final manuscript as submitted.
– Dr Gomes conceptualized and designed the study, contributed to literature search and analysis and interpretation of the case, reviewed and revised the manuscript, and approved the final manuscript as submitted.
– Dr Correia contributed to the interpretation of the case, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Received for publication: Aug 30, 2016
Accepted in revised form: Aug 4, 2017

