










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.31 no.4 Lisboa dez. 2017
REVIEW ARTICLE
Lecithin-cholesterol acyltransferase deficiency: a review for clinical nephrologists
Rute Carmo1, I. Castro-Ferreira1,2, João Paulo Oliveira2,3
1 Nephrology Department, Centro Hospitalar São João, Porto
2 I3S – Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Porto
3 Medical Genetics Department and Reference Center for Hereditary Metabolic Diseases, Centro Hospitalar São João, Porto
ABSTRACT
Lecithin-cholesterol acyltransferase (LCAT) is the enzyme responsible for esterification of free cholesterol on the surface of lipoproteins, particularly in high-density lipoproteins (HDL), and is also involved in the reverse transport of cholesterol from peripheral tissues to the liver. LCAT is synthesized in the liver and circulates in plasma reversibly bound to lipoprotein particles, or in a lipid-free form.
Primary LCAT deficiency is a rare inherited metabolic condition caused by homozygous or compound heterozygous mutation in the LCAT gene. It is associated with two distinct clinical syndromes, Familial LCAT Deficiency (FLD) and Fish-Eye Disease (FED), respectively caused by complete and partial deficiency of LCAT activity, but having in common markedly reduced plasma levels of HDL and apolipoprotein A-I. FLD is characterized by corneal opacities, haemolytic anaemia and chronic kidney disease (CKD), which may progress to end-stage renal disease (ESRD). The pathogenesis of FLD nephropathy is not clear, but accumulation of lipoprotein-X in the kidneys might be a major contributing factor. Corneal opacification is the only clinical hallmark of FED.
In line with several reports of intermediate phenotypes, we have recently described an incomplete FLD phenotype in two Portuguese brothers, homozygous for a novel LCAT mutation, presenting with proteinuric CKD but no haemolytic anaemia, who developed noticeable corneal clouding only many years afterwards. Such a phenotype poses a diagnostic challenge to nephrologists, which will have to rely on accurate appraisal of the lipid profile abnormalities and a high index of suspicion to consider the right diagnosis.
Further studies are needed to confirm whether recombinant human LCAT is effective in halting CKD progression in FLD patients. Meanwhile, renoprotective therapy by inhibition of renin-angiotensin-aldosterone system should be initiated as soon as possible. Despite early histological recurrence of the nephropathy in kidney grafts, renal transplantation remains a suitable therapy for FLD patients with ESRD.
Keywords: Familial lecithin-cholesterol acyltransferase deficiency; Fish-eye disease; corneal clouding; chronic kidney disease; renal transplantation; recombinant human LCAT
INTRODUCTION
Lecithin-cholesterol acyltransferase (LCAT) is the enzyme responsible for the esterification of free cholesterol on the surface of lipoproteins, particularly in high density lipoproteins (HDL)1. It is also involved in the reverse transport of cholesterol, a metabolic pathway that mediates the removal of excess cholesterol from peripheral tissues (including macrophages in the arterial wall), and its hepatic delivery for biliary excretion, via the plasma compartment2,3.
LCAT is synthesized in the liver and secreted to the plasma where it circulates reversibly bound to lipoprotein particles, or in a lipid-free form. The plasma concentration of LCAT is about 5-6 mg/L, and may vary slightly with age, gender, smoking and dietary habits3,4. LCAT mass levels in plasma are highly correlated with its catalytic activity and the cholesterol esterification rate5.
The LCAT gene (OMIM*606967; http://omim.org/entry/606967) maps to chromosome16q22.1, has a total genomic size of 4.2 kb and contains 6 exons6. The gene encodes for a polypeptide with 416 amino acid residues, containing four N-glycosylation and two O-glycosylation sites6. The molecular weight of LCAT is approximately 67 kDa, with the linked carbohydrates constituting about 25% of the total mass of the enzyme6.
LCAT deficiency is an ultra-rare autosomal recessive inborn error of lipid metabolism, with a worldwide prevalence below 1:1,000,0007. The disease was originally reported in 19678, in a Norwegian female presenting with anaemia, proteinuria, corneal opacities due to lipid deposits, and evidence of renal and bone marrow accumulation of foamy cells. She also had markedly reduced plasma concentration of esterified cholesterol with a high concentration of free cholesterol, and a severe deficiency of α- and pre-β-lipoproteins, associated with undetectable plasma LCAT activity. Two of her sisters were diagnosed with the same disease. Since then, about 60 sporadic cases and 70 families with complete or partial LCAT deficiency have been identified3.
The Human Gene Mutation Database (HGMD®) currently lists 102 functionally relevant LCAT gene variants, including 77 missense/nonsense point mutations (http://www.hgmd.cf.ac.uk; last accessed on December 1, 2017). The pathogenic mutations are dispersed throughout the entire gene and, in most cases, it is not possible to anticipate the phenotype based on the position of the mutation in the polypeptide chain9.
The purpose of this review is to summarize the role of LCAT in the lipoprotein metabolism; describe the clinical syndromes associated with LCAT deficiency, emphasizing the renal involvement; and highlight the recent therapeutic developments.
ROLE OF LCAT IN THE LIPOPROTEIN METABOLISM
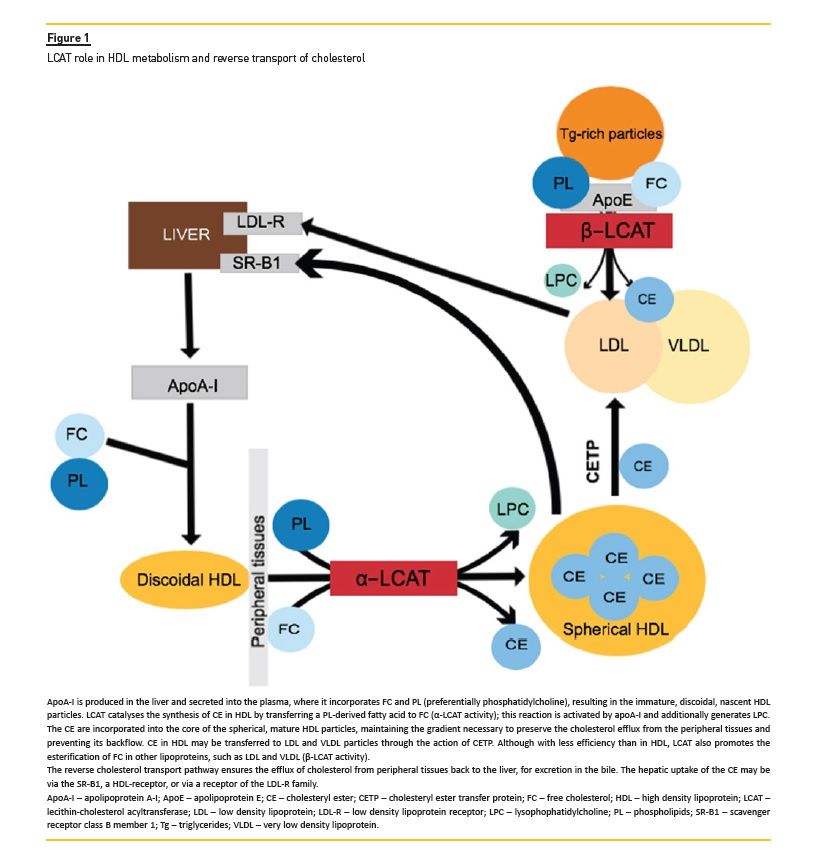
LCAT activity accounts for 90% of the synthesis of cholesterol esters in plasma3. The enzyme reacts mainly with the discoidal, nascent, pre-β1-HDL particles, containing apolipoprotein (apo) A-I, where it esterifies free cholesterol via α-LCAT activity4 (Figure 1). This esterification process consists of the transfer of the sn-2 fatty acyl group of phosphatidylcholine to the 3-β hydroxyl group of free cholesterol, forming a cholesteryl ester and lysophosphatidylcholine3. The cholesteryl esters are then incorporated into the HDL particles, resulting in the formation of the spherical, mature, α-migrating forms of HDL4. In addition to the α-LCAT activity, the enzyme additionally exhibits β-LCAT activity in apoBcontaining lipoproteins, such as low density lipoprotein (LDL) and very low density lipoprotein (VLDL), where apoE is the enzymatic cofactor. LCAT may also be activated, albeit with less efficiency, by other apolipoproteins, such as apoA-II, apoA-IV and apoCI-III3.
The contribution of LCAT to the reverse cholesterol transport occurs either directly, by interaction of mature HDL with the hepatic scavenger receptor class B member 1 (SR-B1), or indirectly, via the cholesteryl ester transfer protein (CETP)-mediated pathway (Figure 1).
The latter pathway promotes the transfer of esterified cholesterol in HDL to the apoB-containing lipoproteins, which are mostly cleared from the circulation through LDL receptor-mediated endocytosis in the liver3.
LCAT DEFICIENCY SYNDROMES
Familial LCAT Deficiency (FLD or Norum disease, OMIM#245900; http://omim.org/entry/245900) and Fish Eye Disease (FED, OMIM#136120; http://omim.org/entry/136120) are the two major syndromes associated to LCAT mutations, but intermediate phenotypes have also been described4,9–11. As LCAT deficiency is invariably associated with markedly reduced HDL and apoA-I levels, the lipid profile is not useful to distinguish the different clinical phenotypes of the disease10. Furthermore, genotype-phenotype correlations have been difficult to establish, since affected relatives may have different clinical and biochemical manifestations10.
Cases of acquired LCAT deficiency due to the development of neutralizing auto-antibodies targeting the enzyme have also been reported, making LCAT genotyping critical for the differential diagnosis between the inherited and the acquired forms of the disease12,13.
Familial LCAT deficiency (FLD)
FLD develops when there is loss of both α-and β-LCAT activities, due to LCAT mutations that impair its synthesis or the hepatic secretion, or result in the production of a severely defective enzyme14. Typically, these patients present with markedly diminished plasma levels of HDL (< 20 mg/dL), have low plasma levels of total and esterified cholesterol, and raised plasma concentrations of free cholesterol, triglycerides and phospholipids3,10,14,15. The ratio of free-to-total cholesterol, which normally is around 30%, increases more than 2.5-fold in patients with FLD; conversely, the esterified-to-free cholesterol ratio is extremely reduced.
The plasma levels of LDL are decreased, as a result of the accelerated catabolism of these lipoproteins, and the concentration of VLDL is increased. The plasma levels of apoA-I and apoA-II are low, while the levels of apoE are often increased.
Bilateral, progressive corneal clouding, haemolytic anaemia, and progressive, proteinuric chronic kidney disease (CKD) are the major clinical complications of FLD8,14.
Lipid deposition in the cornea is responsible for the corneal clouding16,17. It consists of numerous, minute, greyish dot-like opacities forming a mosaic pattern, scattered throughout the corneal stroma but being especially prominent near the limbal area, resembling arcus senilis18. The corneal opacities usually appear early in life and develop gradually over time, constituting the first noticeable sign of the disease in the majority of patients10,14. On slit-lamp examination of the cornea, the lipid dots can be seen in all layers of the corneal parenchyma. Light microscopy shows numerous small vacuoles dispersed in all the histological layers of the cornea, but most densely in the anterior stroma; on electron microscopy study, some of these vacuoles contain electron-dense material17. In vivo confocal microscopy is useful for the differential diagnosis with other metabolic corneal dystrophies, since it confirms the presence of diffuse dark striae and hyperreflective deposits, corresponding to excessive extracellular deposition of cholesterol17. The corneal opacification may be severe enough to cause visual impairment, sometimes requiring corneal transplantation16,18.
Haemolytic, normocytic normochromic anaemia, characteristically of low clinical severity, is caused by deposition of free cholesterol and phosphatidylcholine in the membrane of the red blood cells, shortening their lifespan. Bone marrow biopsy may show foam cells and sea-blue histiocytes, presumably due to the excess of intracellular storage of lipids14.
CKD is the principal cause of morbidity and mortality in patients with FLD14,19. The presenting manifestation of the kidney involvement is proteinuria, which is usually first detected during childhood or adolescence, often reaching nephrotic levels. The urine sediment examination frequently reveals micro hematuria and hyaline casts20. In many patients, CKD progresses to end-stage renal disease (ESRD) by the 4th or 5th decade14,19.
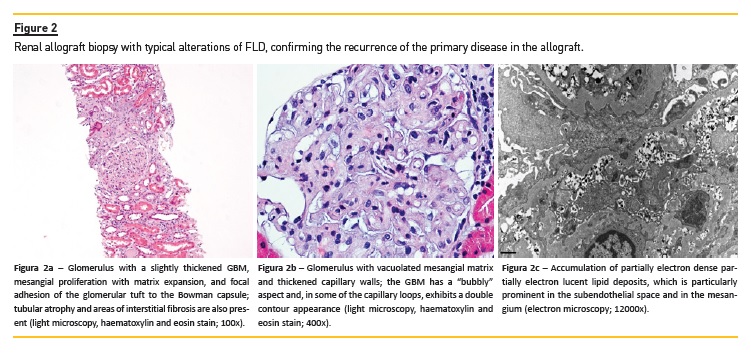
In early stages, renal histopathological findings on light microscopy may resemble membranous nephropathy with mild mesangial expansion, vacuolation of the glomerular capillary walls and, in silver-stained sections, presence of spikes on the glomerular basement membrane (GBM)19,21; the thickening and vacuolation of the GBM, sometimes with double contouring, becomes more prominent as the disease progresses, due to the continuing lipid deposition (Figures 2a, 2b). Vacuolation may also be observed in the Bowman capsule, in the mesangial matrix, as well as in the arterioles and interlobular arteries. On electron microscopy examination, the accumulated lipid appear as small, irregular, partially deeply osmiophilic deposits, containing serpiginous fibrils, rounded lamellar and granular densities19,21; they are particularly numerous in the lamina densa of the GBM, in the subepithelial and subendothelial aspects of the GBM and in the mesangial matrix, but are also present in the Bowman capsule and the vascular endothelium. In rarer cases, intraluminal thrombus-like deposits with a peculiar concentrically lamellated substructure may be seen in dilated glomerular capillary loops (Figure 2c). Over time, these pathological processes eventually lead to the glomerulosclerosis, tubular atrophy and interstitial fibrosis underlying CKD progression.
Although the pathogenesis of renal disease is not entirely understood, renal accumulation of lipoprotein-X (Lp-X) seems to be a major contributing factor15. This hypothesis is supported by the experimental observation that only LCAT-/- knockout (KO) mice with high plasma levels of Lp-X developed renal disease22,23. Lp-X is a multilamellar vesicle enriched in free cholesterol and relatively devoid of cholesteryl esters, triglycerides and apolipoproteins14. In vitro studies showed that Lp-X has cytotoxic and pro-inflammatory properties. It stimulates monocyte infiltration of the glomeruli via a mechanism involving mesangial monocyte chemoattractant protein-1 (MCP-1/CCL2) expression24. Administration of Lp-X to LCAT KO mice results in its accumulation in the kidney, with consequent GBM and endothelial damage, podocyte effacement, expansion of the mesangial matrix and renal tubule vacuolation15. The observation of mesangial C3 staining on the immunofluorescence microscopy study of renal biopsies of patients with FLD20,25 suggest that complement may also have a role in the pathogenesis of the associated nephropathy.
Although accumulation of cholesterol-laden foam cells also occurs in other tissues, the development of hepatomegaly, splenomegaly or lymphadenopathy in patients with FLD has seldom been reported14.
Fish eye disease (FED)
FED is a milder phenotype with predominant corneal involvement, without anaemia or CKD. Since there is only deficiency of α-LCAT activity with preserved β-LCAT activity, the cholesterol esterification rate and the percentage of esterified cholesterol are normal, and there is no Lp-X renal accumulation15,26. As in FLD, the plasma levels of HDL and apoA-I are decreased, and triglycerides are normal to increased10.
Intermediate phenotypes
The diagnosis of FLD may be especially difficult to recognize in patients with atypical phenotypes. We have recently described an incomplete FLD phenotype in two Portuguese brothers apparently homozygous for a novel missense LCAT gene mutation, who presented with CKD and nephrotic proteinuria, progressing to ESRD in the 4th to 5th decade of life11. The histopathological diagnosis of membranous nephropathy in the kidney biopsy of one of the sibs, the absence of haemolytic anaemia, and the late development of corneal opacification, only several years after the start of renal replacement therapy (RRT), significantly delayed the recognition of the underlying metabolic disorder. Such clinical presentations pose a diagnostic challenge to nephrologists, which will have to rely on accurate appraisal of the lipid profile abnormalities and a high index of suspicion in order to arrive at theright diagnosis.
ATHEROSCLEROSIS IN LCAT DEFICIENCY SYNDROMES
Because HDL levels are an inverse predictor of cardiovascular events7, it might be expected that individuals with LCAT deficiency have an increased cardiovascular risk, as observed in other diseases characterized by markedly reduced HDL, such as Tangier disease and apoA-I deficiency27. However, whether LCAT deficiency syndromes are associated with an increased cardiovascular risk is still a controversial issue, despite the numerous studies that have addressed the possible role of LCAT in the pathogenesis of atherosclerosis.
Experimental animal studies have yielded inconsistent results. In most mouse models, overexpression of LCAT was found to be pro-atherogenic despite the significant increase in HDL levels, while studies in rabbits and nonhuman primates indicate that LCAT is likely atheroprotective.
These contradictory outcomes were attributed to the absence of CETP in mice, as opposed to rabbits and humans3,26. Overall, these data suggest that the LCAT role in the pathogenesis of atherosclerosis is dependent on additional factors, such as diet and other genes involved in the reverse cholesterol transport26.
The available evidence from observational human studies addressing the risk of atherosclerosis in LCAT deficiency syndromes is also conflicting4,26. No vascular events or deaths were reported over a 25-year follow-up of a large Canadian kindred with FLD, including two homozygous patients with no residual LCAT activity and 9 heterozygous relatives28, and development of detectable atherosclerotic plaques on carotid ultrasound imaging only occurred in four of the heterozygotes29. A further, particularly interesting, observation is that premature coronary heart disease (CHD) has been more frequently reported in patients diagnosed with FED than in those with FLD9. Other worth mentioning cross-sectional studies that have used the ultrasound measurement of carotid intima-media thickness (cIMT) as a surrogate marker for atherosclerosis have contrastingly reported either a modest increase in average cIMT in heterozygous carriers of LCAT mutations as compared to unaffected subjects30, or a lower average cIMT in carriers than in healthy controls, with the inheritance of a mutated LCAT genotype exerting a remarkable genedose dependent effect in reducing cIMT31, indicating that LCAT deficiency does not enhance preclinical atherosclerosis, despite its notable HDL lowering effect.
These findings are explainable by the accumulation of pre-β-HDL particles in plasma, associated with defective LCAT function, enhancing the efflux of cholesterol from macrophages4, as well as by the low plasma levels of LDL and apoB present in homozygous LCAT-deficient patients26. Further studies in larger cohorts are warranted to clarify the cardiovascular risk associated with genetic LCAT deficiency, and would be of substantial help in defining the role of LCAT in CHD development4.
TREATMENT
Currently, there is no specific treatment available. Dietary changes are recommended, such as restriction of dietary fat, but the efficacy of lipid-lowering medications is still unknown. Additionally, angiotensin II receptors blockers (ARBII) or angiotensin-converting enzyme inhibitors (ACEI) are advisable for patients with proteinuria14.
There is a case report of successful remission of proteinuria using low dose steroids in association with ARBII and ACEI, arguably as a result of the anti-inflammatory and immunosuppressive properties of the steroids, which downregulates the activity of nuclear factor kappa B (NF-kB)20. RRT is required for patients who develop ESRD.
Despite the documentation of early graft recurrence in biopsies performed even in the first weeks after transplantation, renal transplantation is not contraindicated, since there is a favourable long-term graft survival25,32,33. Sequential liver-kidney transplantation from a living donor was attempted as a curative treatment.
In this case, the patient had no recurrence of renal disease, despite the abnormal lipid profile compatible with LCAT deficiency34.
A recombinant human LCAT (rhLCAT) is being developed for the treatment of FLD patients with renal disease.
In LCAT KO mice, injection of rhLCAT resulted in normalization of lipid profile and transformation of Lp-X in HDL-like particles35. In a phase 1b study of patients with CHD and low HDL plasma levels, treatment with rhLCAT has shown acceptable safety and tolerability profiles36. Furthermore, during an 8-month course of experimental rhLCAT treatment in a patient with FLD and advanced CKD there were beneficial effects in several clinical, biochemical and lipoprotein parameters, although the improvement of renal function was only slight and transitory37.
CONCLUSIONS
LCAT is a lipoprotein-associated enzyme which plays a major role in the esterification of free cholesterol, maturation of HDL particles and reverse cholesterol transport. Primary LCAT deficiency is an ultra-rare autosomal recessive metabolic condition, underlying two distinct clinical syndromes, FLD and FED, respectively associated with complete and partial loss of LCAT activity.
Regardless of the degree of residual enzyme activity, low HDL cholesterol levels are a biochemical hallmark of LCAT-deficient patients.
The typical FLD clinical phenotype is characterized by early-manifesting corneal opacities, haemolytic anaemia, and proteinuria, commonly in the nephrotic range. Progressive CKD, eventually reaching ESRD by the 4th or 5th decade, is a major cause of morbidity and mortality in these patients. The pathogenesis of the renal involvement in FLD is not completely understood, but the accumulation of Lp-X on the kidneys may be the most important contributing factor. Renal biopsy remains a critical diagnostic tool: although in its early stages the FLD nephropathy may be difficult to distinguish from membranous nephropathy, based exclusively on the light microscopy findings, electron microscopy will show large numbers of lipid deposits in subepithelial, subendothelial, intramembranous and mesangial locations.
In order to not miss the correct diagnosis, practicing nephrologists should be aware of patients presenting with attenuated or incomplete FLD phenotypes, without haemolytic anaemia but manifesting severe proteinuria and progressive CKD many years before the development of noticeable corneal clouding. In such cases, unexplained low HDL levels are a major clue to the diagnosis, which can be confirmed by molecular genetic testing demonstrating homozygosity or compound heterozygosity for pathogenic variants of the LCAT gene. Furthermore, even in patients on RRT, the development of noticeable corneal clouding should prompt clinicians to consider the diagnosis of FLD.
Despite the low HDL levels, FLD patients do not appear to be at high risk for cardiovascular events.
Further studies are needed to confirm whether rhLCAT is effective in halting renal disease progression, when started at an earlier stage of the disease. Until then, renoprotective therapy by inhibition of renin-angiotensin-aldosterone system should be initiated as soon as possible.
References
1. Glomset JA, Janssen ET, Kennedy R, Dobbins J. Role of plasma lecithin:cholesterol acyltransferase in the metabolism of high density lipoproteins. J Lipid Res.1966;7:638-648. [ Links ]
2. Glomset JA. The plasma lecithin:cholesterol acyltransferase reaction. J Lipid Res. 1968;9(2):155-167. [ Links ]
3. Kunnen S, Van Eck M. Lecithin:cholesterol acyltransferase: old friend or foe in atherosclerosis? J Lipid Res. 2012;53(9):1783-1799. [ Links ]
4. Calabresi L, Simonelli S, Gomaraschi M, Franceschini G. Genetic lecithin: cholesterol acyltransferase deficiency and cardiovascular disease. Atherosclerosis. 2012;222(2):299-306. [ Links ]
5. Albers JJ, Chen CH, Adolphson JL. Lecithin:cholesterol acyltransferase (LCAT) mass; its relationship to LCAT activity and cholesterol esterification rate. J Lipid Res. 1981;22(8):1206-1213. [ Links ]
6. Mclean J, Wion K, Drayna D, Fielding C, Lawn R. Human lecithin-cholesterol acyltransferase gene: complete gene sequence and sites of expression. Nucleic Acids Res. 1986;14(23):9397-9406. [ Links ]
7. Savel J, Lafitte M, Pucheu Y, Pradeau V, Tabarin A, Couffinhal T. Very low levels of HDL cholesterol and atherosclerosis, a variable relationship – a review of LCAT deficiency. Vasc Health Risk Manag. 2012;8(1):357-361. [ Links ]
8. Norum KR, Gjone E. Familial plasma lecithin: cholesterol acyltransferase deficiency. biochemical study of a new inborn error of metabolism. Scand J clin Lab Invest. 1967;20(8):231-243. [ Links ]
9. Kuivenhoven J a, Pritchard H, Hill J, Frohlich J, Assmann G, Kastelein J. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res. 1997;38(2):191-205.
10. Calabresi L, Pisciotta L, Costantin A, et al. The molecular basis of lecithin: cholesterol acyltransferase deficiency syndromes: a comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler Thromb Vasc Biol.> 2005;25(9):1972-1978. [ Links ]
11. Castro-Ferreira I, Carmo R, Silva SE, et al. Novel missense LCAT gene mutation associated with an atypical phenotype of familial LCAT deficiency in two portuguese brothers. JIMD Rep. 2017;4:113-116. [ Links ]
12. Takahashi S, Hiromura K, Tsukida M, et al. Nephrotic syndrome caused by immunemediated acquired LCAT deficiency. J Am Soc Nephrol. 2013;24(8):1305-1312. [ Links ]
13. Akiko T, Okura T, Nagao T, et al. A case of acquired lecithin:cholesterol acyltransferase deficiency with sarcoidosis that remitted spontaneously. CEN case reports. 2016;5(2):192-196. [ Links ]
14. Saeedi R, Li M, Frohlich J. A review on lecithin:cholesterol acyltransferase deficiency. Clin Biochem. 2015;48(7-8):472-475. [ Links ]
15. Ossoli A, Neufeld EB, Thacker SG, et al. Lipoprotein X causes renal disease in LCAT deficiency. PLoS One. 2016;11(2):1-26. [ Links ]
16. Izzedine H, Bodaghi B, Launay-Vacher V, Deray G, Editor F. Eye and kidney: drom clinical findings to genetic explanations. J Am Soc Nephrol. 2003;14(2):516-529. [ Links ]
17. Palmiero P-M, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin-cholesterol acyltransferase deficiency. Cornea. 2009;28(9):1061-1064. [ Links ]
18. Schaefer EJ, Santos RD, Asztalos BF. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol. 2010;21(4):289-297. [ Links ]
19. Najafian B, Lusco MA, Finn LS, Alpers CE, Fogo AB. AJKD atlas of renal pathology: lecithin–cholesterol acyltransferase (LCAT) feficiency. Am J Kidney Dis. 2017;69(3):e5-e6. [ Links ]
20. Miarka P, Idzior-Waluś B, Kuźniewski M, Waluś-Miarka M, Klupa T, Sułowicz W. Corticosteroid treatment of kidney disease in a patient with familial lecithin-cholesterol acyltransferase deficiency. Clin Exp Nephrol. 2011;15(3):424-429. [ Links ]
21. Hirashio S, Ueno T, Naito T, Masaki T. Characteristic kidney pathology, gene abnormality and treatments in LCAT deficiency. Clin Exp Nephrol. 2014;18(2):189-193. [ Links ]
22. Lambert G, Sakai N, Vaisman BL, et al. Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J Biol Chem. 2001;276(18):15090-15098. [ Links ]
23. Zhu X, Herzenberg AM, Eskandarian M, et al. A novel in vivo lecithin-cholesterol acyltransferase (LCAT)-deficient mouse expressing predominantly LpX is associated with spontaneous glomerulopathy. Am J Pathol. 2004;165(4):1269-1278. [ Links ]
24. Lynn EG, Siow YL, Frohlich J, et al. Lipoprotein-X stimulates monocyte chemoattractant protein-1 expression in mesangial cells via nuclear factor-KB. Kidney Int. 2001;60(2):520-532. [ Links ]
25. Panescu V, Grignon Y, Hestin D, et al. Nephrology dialysis transplantation recurrence of lecithin cholesterol acyltransferase deficiency after kidney transplantation. 1997:2430-2432. [ Links ]
26. Rousset X, Shamburek R, Vaisman B, Amar M, Remaley AT. Lecithin cholesterol acyltransferase: an anti-or pro-atherogenic factor? Curr Atheroscler Rep. 2011;13(3):249-256. [ Links ]
27. Schaefer EJ, Anthanont P, Asztalos BF. High-density lipoprotein metabolism, composition, function, and deficiency. Curr Opin Lipidol. 2014;25(3):194-199. [ Links ]
28. Frohlich J, McLeod R, Pritchard PH, Fesmire J, McConathy W. Plasma lipoprotein abnormalities in heterozygotes for familial lecithin:cholesterol acyltransferase deficiency. Metabolism. 1988;37(1):3-8. [ Links ]
29. Ayyobi AF, McGladdery SH, Chan S, John Mancini GB, Hill JS, Frohlich JJ. Lecithin: cholesterol acyltransferase (LCAT) deficiency and risk of vascular disease: 25 year follow-up. Atherosclerosis. 2004;177(2):361-366. [ Links ]
30. Hovingh GK, Hutten BA, Holleboom AG, et al. Compromised LCAT function is associated with increased atherosclerosis. Circulation. 2005;112(6):879-884. [ Links ]
31. Calabresi L, Baldassarre D, Castelnuovo S, et al. Functional lecithin: cholesterol acyltransferase Is not required for efficient atheroprotection in humans. Circulation. 2009;120(7):628-635. [ Links ]
32. Strøm EH, Sund S, Reier-Nilsen M, Dørje C, Leren TP. Lecithin: Cholesterol acyltransferase (LCAT) deficiency: renal lesions with early graft recurrence. Ultrastruct Pathol. 2011;35(3):139-145. [ Links ]
33. Liew H, Simpson I, Kanellis J, Mulley WR. Recurrent glomerulopathy in a renal allograft due to lecithin cholesterol acyltransferase deficiency. Nephrology. 2016;21(1):73-74. [ Links ]
34. Ahmad SB, Miller M, Hanish S, et al. Sequential kidney–liver transplantation from the same living donor for lecithin cholesterol acyl transferase deficiency. Clin Transplant. 2016;30(10):1370-1374. [ Links ]
35. Rousset X, Vaisman B, Auerbach B, et al. Effect of recombinant human lecithin cholesterol acyltransferase infusion on lipoprotein metabolism in mice. Lipids. 2010:140-148. [ Links ]
36. Shamburek RD, Bakker-Arkema R, Shamburek AM, et al. Safety and tolerability of ACP-501, a recombinant human lecithin:cholesterol acyltransferase, in a phase 1 single-dose escalation study. Circ Res. 2016;118(1):73-82. [ Links ]
37. Shamburek RD, Bakker-Arkema R, Auerbach BJ, et al. Familial lecithin:cholesterol acyltransferase deficiency: first-in-human treatment with enzyme replacement. J Clin Lipidol. 2016;10(2):356-367. [ Links ]
Rute Carmo, MD
Centro Hospitalar de São João
Alameda Professor Hernâni Monteiro,4200-319 Porto
E-mail: rute.carvalho.carmo@gmail.com
ACKNOWLEDGMENTS
We would like to thank Dr. Pedro Rodrigues-Pereira and Dr. Susana Sampaio for the renal allograft biopsy photomicrographs.
Disclosure of potential conflicts of interest: none declared
Received for publication: Dec 21, 2017
Accepted in revised form: Dec 27, 2017


{kind=link}
{kind=link}