










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.31 no.3 Lisboa set. 2017
CASE REPORT
Atypical renal presentation of antiphospholipid Syndrome
Ana Gaspar1, Rita Manso2, Fernando Pereira1, Liliana Cunha1, Luís Inchaustegui1, Adelaide Serra1, Bruno Rodrigues1, Pedro Correia1
1 Department of Nephrology, Hospital Professor Doutor Fernando da Fonseca
2 Department of Pathology, Hospital Professor Doutor Fernando da Fonseca
ABSTRACT
Antiphospholipid syndrome (APS) is a systemic autoimmune disease which can occur as a primary disease or in association with other autoimmune diseases, the most frequent being Systemic Lupus Erythematosus (SLE). Although renal manifestations of SLE are well known, antiphospholipid syndrome renal manifestations such as antiphospholipid syndrome nephropathy and glomerulopathies have yet to be better characterized. The authors present the case of a 39‑year‑old Caucasian woman with antiphospholipid syndrome diagnosis and a previous history of deep venous thrombosis and intermittent polyarthralgia, who was referred to a nephrology consultation for proteinuria and microscopic haematuria with preserved renal function. The renal biopsy showed a pattern of membranous glomerulopathy and thrombotic microangiopathy in association with antiphospholipid syndrome nephropathy.
This case report illustrates a complex clinical and anatomopathological case of a 39‑ year‑old woman with a previous antiphospholipid syndrome diagnosis who presented with unspecific manifestations such as proteinuria and microscopic haematuria and preserved renal function. The histological findings alert us to the range of possible renal manifestations of APS and the need to better characterize these patients by preforming renal biopsy.
Key‑words: Antiphopholipid Antibodies (aPL); Antiphospholipid Syndrome (APS); Antiphospholipid Syndrome Nephropathy (APSN); Membranous Glomerulonephritis; Renal Biopsy; Systemic Lupus Erythematosus (SLE).
INTRODUCTION
Antiphospholipid syndrome is a systemic autoimmune disease characterized by the presence of circulating antiphospholipid antibodies (aPL). The first international consensus statement on diagnostic criteria of APS was developed in 1998 in Japan and was updated in 2006. The diagnostic criteria include existence of these antibodies and clinical criteria such as thrombotic events in any organ and/or pregnancy morbidity1. It can occur as a primary disease or in association with other autoimmune diseases, the most frequent being Systemic Lupus Erythematosus (SLE). About 30 to 40% of SLE patients have circulating aPL and about half of them will develop APS in a 10 to 20 year follow‑up2.
Both can affect the kidney independently or simultaneously, making the differential diagnosis between inflammatory and thrombotic damage a challenge, as it impacts the way these patients are treated.
APS has been associated with various forms of kidney injury, including large renal vessel thrombosis and renal artery stenosis, but the advent of renal biopsies and renal histopathology has allowed the description and characterization of the microscopic renal damage by the aPL. The spectrum of these alterations, which are associated with deleterious impact on kidney function, has been defined as antiphospholipid nephropathy (APSN). It refers to the ischaemic damage (without inflammatory damage) of the renal parenchyma caused by thrombotic microangiopathy and proliferative and fibrotic lesions of the intra‑renal vessels without inflammatory damage2,3.
The clinical manifestations are unspecific and can range from hypertension, reduced kidney function, proteinuria usually <1.5g/day and microscopic haematuria to acute kidney injury. APSN prevalence in primary APS is estimated to be 2.7% to 9% though it is probably underestimated. In secondary APS associated with SLE, it can range from 9% to 40% due to the fact that biopsies are preformed more consistently in this group and it can coexist with lupus nephritis4.
Moreover, as few kidney biopsies of APS patients are performed, due to the fact that these patients frequently have low platelet counts and are anticoagulated, other kinds of renal manifestations associated with APS besides APSN, namely glomerulopathies have not been well characterized.
The diagnosis of APSN and APS‑associated glomerulopathies is challenging, particularly in patients with diagnosis of SLE, and requires renal biopsy.
CASE REPORT
A 39‑year‑old Caucasian woman, with APS diagnosis two years prior, was referred to a nephrology consultation for proteinuria and microscopic haematuria with preserved renal function. She had a history of deep venous thrombosis of her left leg at the age of 22 and intermittent polyarthralgia complaints of distal joints from the age of 24 to the present.
Due to her medical history, she started attending rheumatology consultations when she was 29 years old. She was prescribed aspirin and corticoids, but did not comply. Four years later a laboratory analysis detected B2‑glicoprotein, anti‑cardiolipin (IgG and IgM) and anti‑SLC70 antibodies, lupic anticoagulant and ANAs without the presence of anti‑Double Stranded DNA (DsDNA) antibodies. Due to patients complaints of migraine, an MRI was performed, which detected a left hemisphere deep vascular ischaemic lesion of the deep branches of the left cerebral anterior artery.
At the age of 39, she was referred to a nephrology consultation. Of her laboratory evaluation, we emphasize urine analysis with proteinuria (++) and haematuria (++), measured 24h proteinuria of 1.7–2.4g/day, sCr 0.7mg/dL and identified B2‑glicoprotein, anti‑cardiolipin and lupic anticoagulant antibodies. The protein electrophoretic pattern was within normal range and immunoglobulin and serum light free chain had no relevant alterations.
The physical exam did not reveal any relevant abnormalities such as hypertension, oedema or neurologic deficits. The laboratory workup showed urine albumin‑creatinine ratio of 2000mg/g, normal complement measurements (C3, C4, CH50), negative DsDNA and the presence of ANAs and anti‑centromere antibodies.
HIV, HBV and HCV serologies were negative. The remaining results were similar. A renal biopsy was preformed and the patient was discharged and treated with ACE inhibitors, hydroxychloroquine and oral anticoagulants (dabigatran).
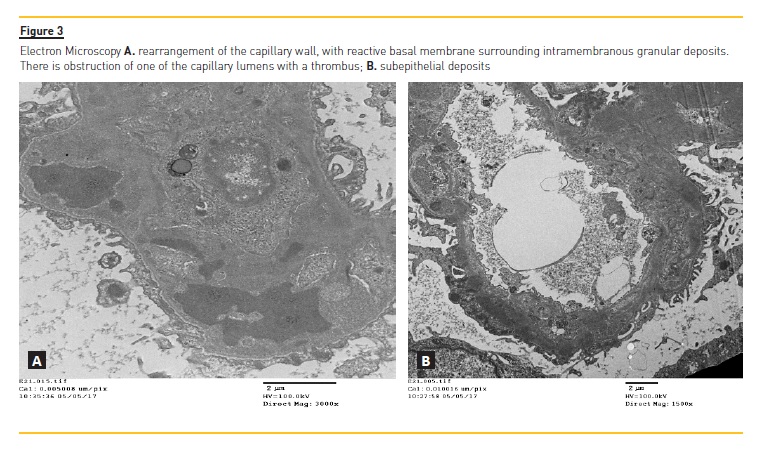
Two weeks later the renal biopsy report described as part of the light microscopy exam membranous glomerulonephritis, most likely secondary, with enlarged glomeruli, global thickening of the capillary walls and a slight segmental expansion of the mesangial matrix without a significant cellularity increase (Fig.1). The silver stain showed countless inclusions (Fig.2). It also described slight double contours on two glomeruli and some hyaline thrombi suggesting chronic ischaemia, which can be present in APS/APSN. The immunofluorescence exam showed IgG, C3 and light chain lambda and kappa universal granular deposits (capillary wall and mesangium), with absence of IgM, IgA, C1, C4 and fibrin. Electron microscopy (Fig. 3 A and 3B) showed an atypical membranous pattern with mesangial, subepithelial and intramembranous granular deposits and a capillary thrombus probably in probable association with APS. Since her discharge, the patient has not returned for follow‑up appointments.
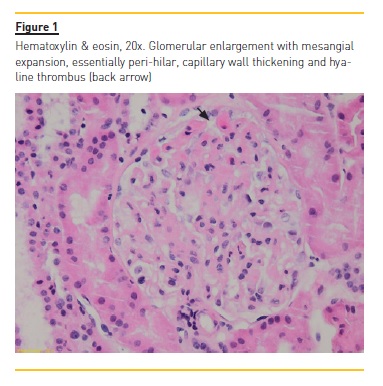
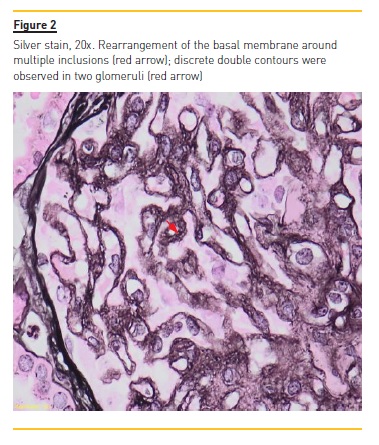
DISCUSSION
This case report illustrates a complex clinical and anatomopathological case of a 39‑year‑old woman with a previous APS diagnosis who presented with unspecific manifestations of proteinuria and microscopic haematuria with preserved renal function.
The patient had a definite APS diagnosis as she presented with persistent aPL longer than 12 weeks, a venous thrombosis episode and a probable arterial cerebral ischaemic lesion documented on the MRI. The hypothesis of SLE in association with APS was suspected based on the presence of three of the eleven American College of Rheumatology (ACR) criteria. These included immunological disorder with the presence of anti‑cardiolipin antibodies, a positive test for antinuclear antibodies (ANA) and persistent proteinuria.
Although there was a description of intermittent polyarthalgia, the patient did not present with erythema or swelling of the joints consistent with a definite diagnosis of non‑erosive arthritis, which would be the fourth criteria necessary for definite SLE diagnosis according to the ACR criteria. According to the Systemic Lupus International Collaborating Clinics group which reviewed and validated the ACR SLE classification criteria in 2012, to establish the diagnosis of SLE the patient has to meet four of the eleven ACR or lupus nephritis criteria documented on a renal biopsy with ANA or anti‑dsDNA antibodies5,6. Considering that the patient only met three ACR criteria, including persistent proteinuria, and there was no evidence of other important aspects such as constitutional symptoms, presence of DsDNA antibodies or low C3 or C4, a renal biopsy was important not only to document renal manifestations of APS but also to assess for lupus nephritis7.
The anatomopathological exam was interpreted as atypical secondary membranous glomerulopathy and thrombotic microangiopathy with thrombi probably associated with ASP/ASPN. The presence of mesangial deposits on electron microscopy made the diagnosis of primary membranous glomerulonephritis less likely, although measurements of anti‑phospholipase A2 receptor antibodies are crucial to definitely exclude this diagnosis. It also did not fulfill diagnostic criteria for type V membranous lupus nephritis due to the absence of hypercelularity and C1q or other immunoglobulin deposits on immunofluorescence exam8,9.
Also, she scored 0 on the point system used to determine both lupic nephritis activity and chronicity indices.
Association of membranous glomerulonephritis with APS is rare, but was described in a series of 29 biopsies performed on APS patients in two university hospitals in Paris in 20033. Of these 29 cases, 69% revealed vascular characteristics of APSN and 31% pathological findings other than APSN. In this last group, the most frequent was membranous nephropathy, representing 33% within this group and 10% of the total10. Other causes of secondary membranous nephropathy, such as malignancies and other autoimmune conditions, must also be excluded in this patients follow‑up.
Another challenging aspect of the anatomopathological exam is the presence of few subtle double contours which in association with peri‑hilar capillary wall mesangial expansion was interpreted as being chronic ischaemic alterations, but poses the hypothesis of a membranoproliferative glomerulonephritis. This hypothesis appeared less probable due to the absence of subendothelial deposits on electron microscopy9,10.
The initial treatment with hydroxychloroquine and ACE inhibitors took into account the possible SLE diagnosis in association with subnephrotic proteinuria in an otherwise asymptomatic patient. However the authors concluded that based on the biopsy findings, the patient did not meet criteria for a definite SLE. As the patient did not return for follow‑up appointments, the treatment and its outcome has yet to be assessed.
For the APS diagnosis, this patient started treatment with oral anticoagulants. Although the approved anticoagulants for this condition consist of vitamin K antagonists, preliminary trials supporting the use of new oral anticoagulants have recently been reported3,11. Due to this patient previously showing non‑compliance with her medication, dabigatran was decided on as treatment since it has proven to be safe in patients with venous thrombosis history, without the need for laboratory monitoring. The patient was informed and agreed to this treatment.
This case illustrates the complexity of renal manifestations of APS and demonstrates the importance of renal biopsy and anatomopathological exam, including electron microscopy, to better characterize these patients.
References
1. Miyakis S, Lockshin MD, Atsumi T, et al. International Consensus statement on an update of the classification criteria for definite antiphospholipid syndrome. J Thromb Haemost.2006;4(2):295‑306 [ Links ]
2. Siascia S, Baldovino S, Schreiber K, Solfietti L, Roccatello D. Antiphospholipid syndrome and the kidney. Semin in Nephrol. 2015;35(5):478‑486 [ Links ]
3. Bienaimé F, Legendre C, Terzi F, Canaud G. Antiphospholipid syndrome and kidney disease. Kidney Int. 2017;91(1):34‑44 [ Links ]
4. Fakhouri F, Noel L, Zuber J, et al. The expanding spectrum of renal diseases associated with antiphospholipid syndrome. Am J Kidney Dis. 2003;41(6):1205‑1211 [ Links ]
5. Almaani S, Meara A, Rovin B. Update on lupus nephritis. Clin J Am Soc Nephrol. 2017;12(5):825‑835 [ Links ]
6. Petri M, Orbai A, Alarcón GS. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677‑2686 [ Links ]
7. Hahn B, MacMahon M, Wilkinson A, et al. American college of reumathology guidelines for the screening, treatment and management of lupus nephritis. Arthritis Care Res. 2012;64(6):797–808 [ Links ]
8. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO Clinical practice guidelines for glomerulonephritis. Kidney Int Suppl. 2012;2(2):139‑274 [ Links ]
9. Sethi S, Haas M, Markowitz G, et al. Mayo Clinic/Renal Pathology Society consensus report on pathologic classification, diagnosis and reporting of GN. J Am Soc Nephrology. 2015;27(5): 1278‑1287 [ Links ]
10. Yang AH, Lin BS, Kuo KL, et al. The clinicopathological implications of endothelial tubuloreticular inclusions found in glomeruli having histopathology of idiopathic membranous nephropathy. Nephrol Dial Transplant. 2009; 24(11):3419‑3425 [ Links ]
11. Siascia S, Breen K, Hunt BJ. Rivaroxaban use in patients with antiphospholipid syndrome and previous venous thomboembolism. Blood Coagul Fibrinolysis. 2015;26(4):476‑477 [ Links ]
Ana Gaspar, MD
Department of Nephrology of Hospital Professor Doutor Fernando da Fonseca
E‑mail: ana.ccs.gaspar@gmail.com
Disclosure of potential conflicts of interest: none declared.
Received for publication: Jun 19, 2017
Accepted in revised form: Sep 20, 2017<


{kind=link}